Department of Biological Sciences, Ulsan National Institute of Science and Technology, Ulsan, 44919, Republic of Korea.
Department of Genetics, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, 19104, USA.
Nat Commun. 2023 Mar 21;14(1):1570. doi: 10.1038/s41467-023-37126-3.
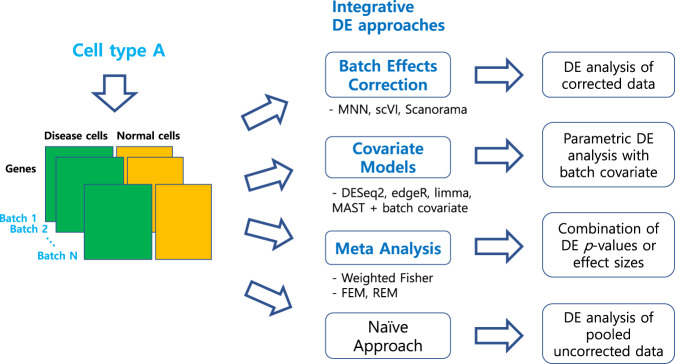
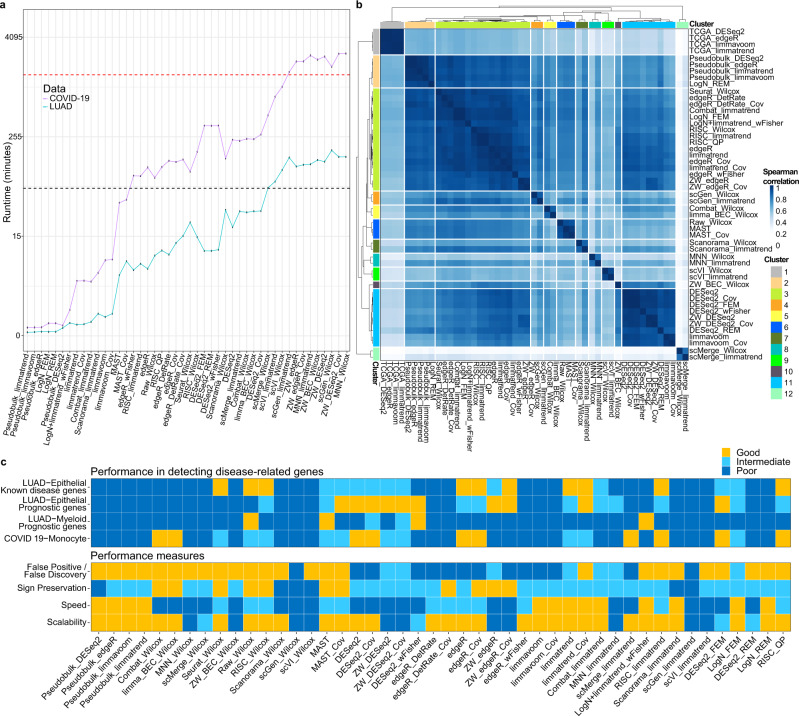
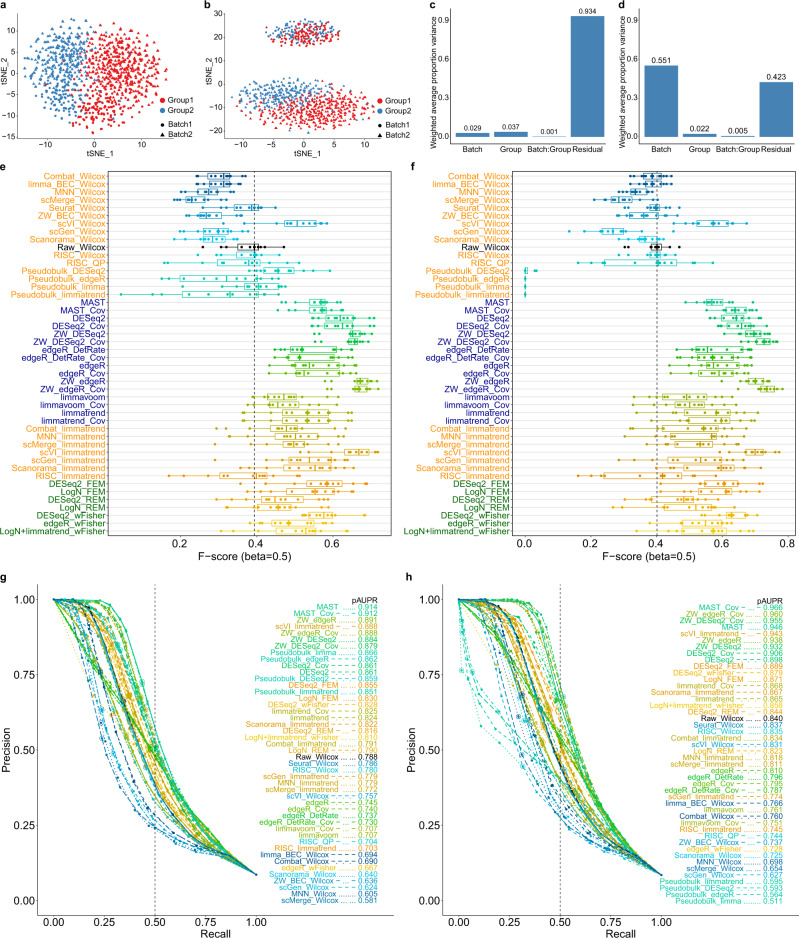
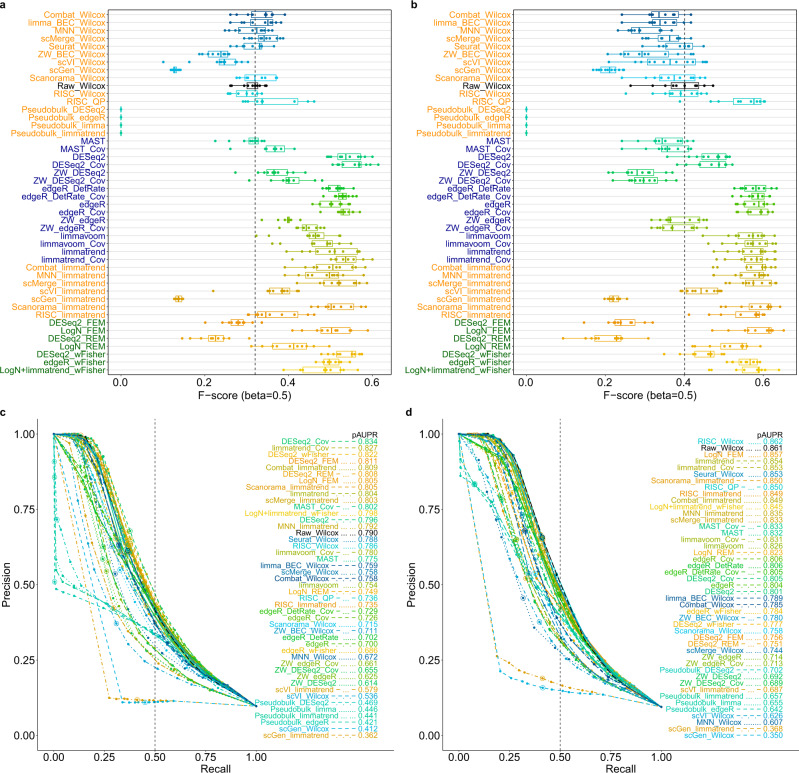
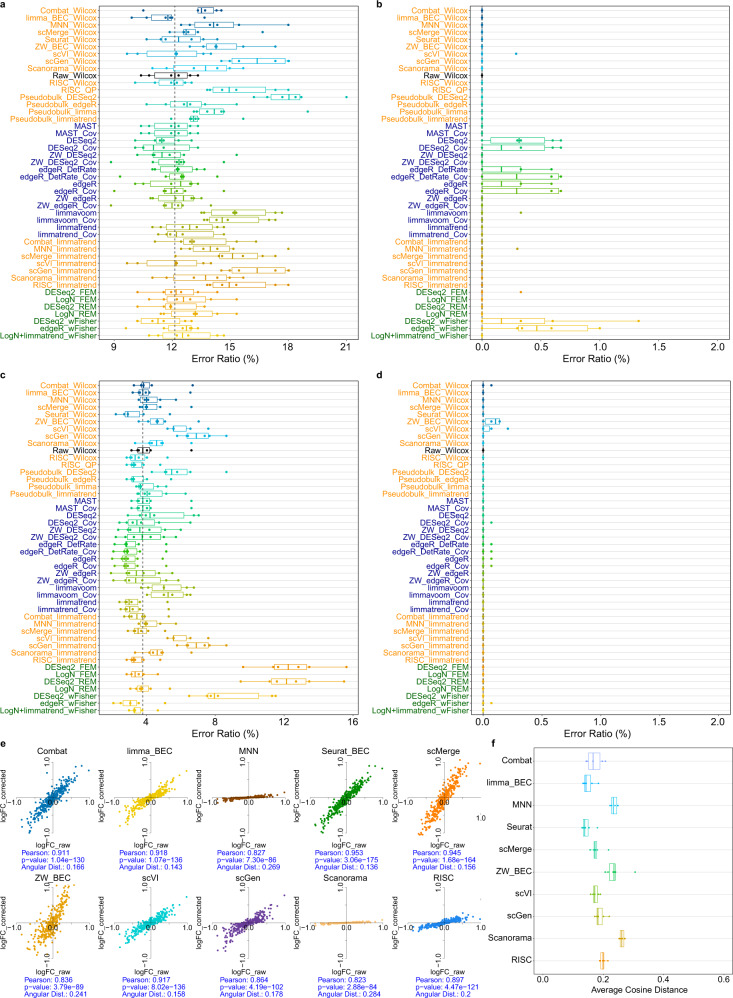
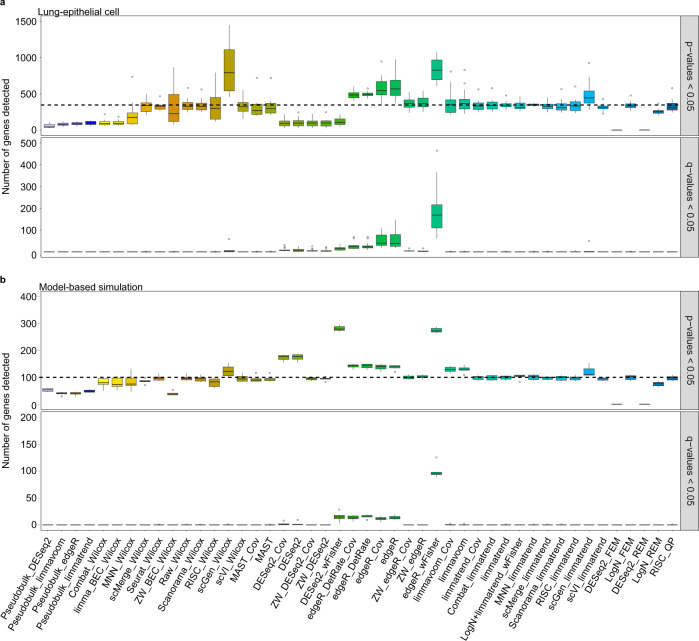
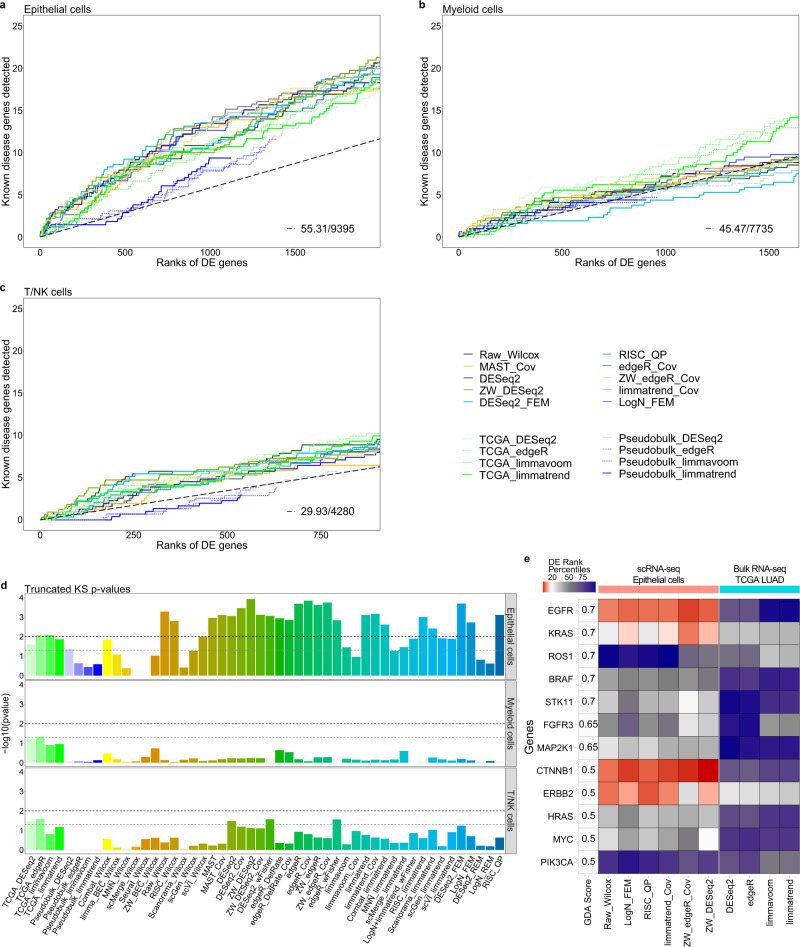
Integration of single-cell RNA sequencing data between different samples has been a major challenge for analyzing cell populations. However, strategies to integrate differential expression analysis of single-cell data remain underinvestigated. Here, we benchmark 46 workflows for differential expression analysis of single-cell data with multiple batches. We show that batch effects, sequencing depth and data sparsity substantially impact their performances. Notably, we find that the use of batch-corrected data rarely improves the analysis for sparse data, whereas batch covariate modeling improves the analysis for substantial batch effects. We show that for low depth data, single-cell techniques based on zero-inflation model deteriorate the performance, whereas the analysis of uncorrected data using limmatrend, Wilcoxon test and fixed effects model performs well. We suggest several high-performance methods under different conditions based on various simulation and real data analyses. Additionally, we demonstrate that differential expression analysis for a specific cell type outperforms that of large-scale bulk sample data in prioritizing disease-related genes.
单细胞 RNA 测序数据在不同样本之间的整合一直是分析细胞群体的主要挑战。然而,单细胞数据差异表达分析的整合策略仍未得到充分研究。在这里,我们对 46 种用于多批次单细胞数据差异表达分析的工作流程进行了基准测试。我们表明,批次效应、测序深度和数据稀疏度极大地影响了它们的性能。值得注意的是,我们发现使用经过批次校正的数据很少能改善稀疏数据的分析,而对大量批次效应进行批次协变量建模则可以改善分析。我们表明,对于低深度数据,基于零膨胀模型的单细胞技术会降低性能,而使用 limmatrend、Wilcoxon 检验和固定效应模型对未经校正的数据进行分析则表现良好。我们根据各种模拟和真实数据分析,在不同条件下提出了几种高性能方法。此外,我们证明,针对特定细胞类型的差异表达分析在优先考虑与疾病相关的基因方面优于大规模批量样本数据。