Latvian Biomedical Research and Study Centre, Riga, Latvia.
Riga East Clinical University Hospital, Centre of Tuberculosis and Lung Diseases, Stopiņi Region, Upeslejas, Latvia.
Microb Genom. 2023 Mar;9(3). doi: 10.1099/mgen.0.000956.
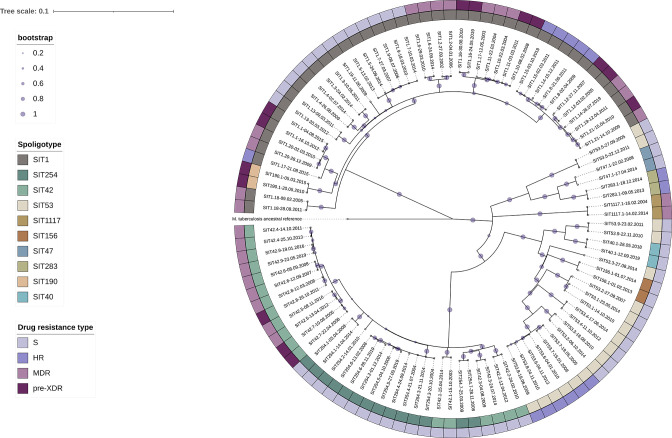
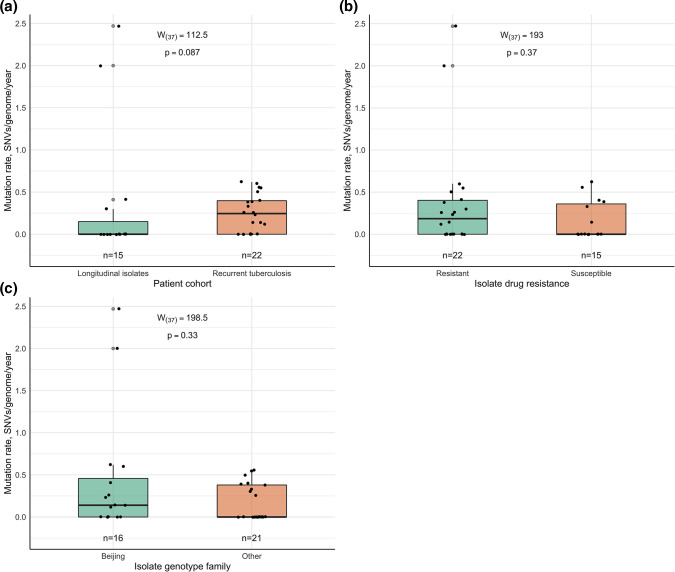
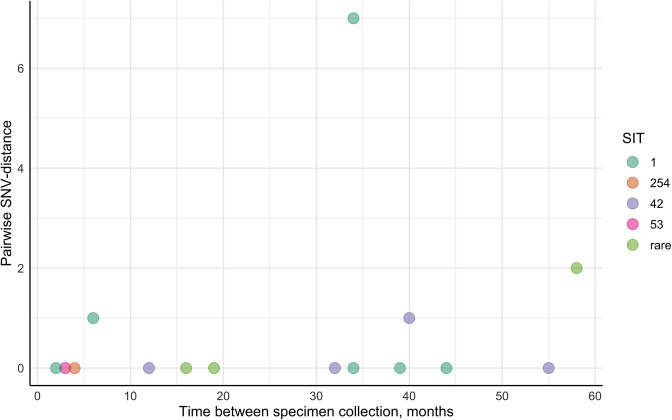
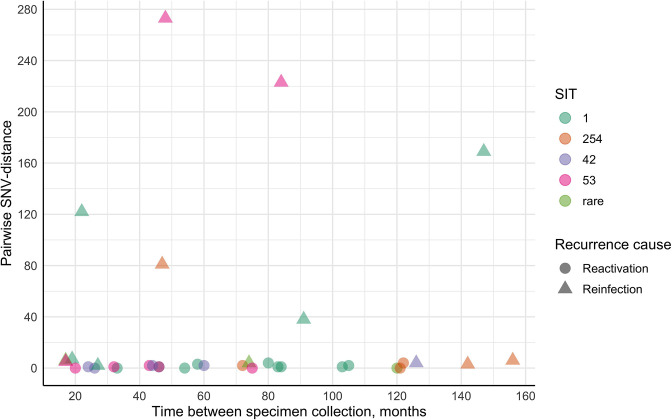
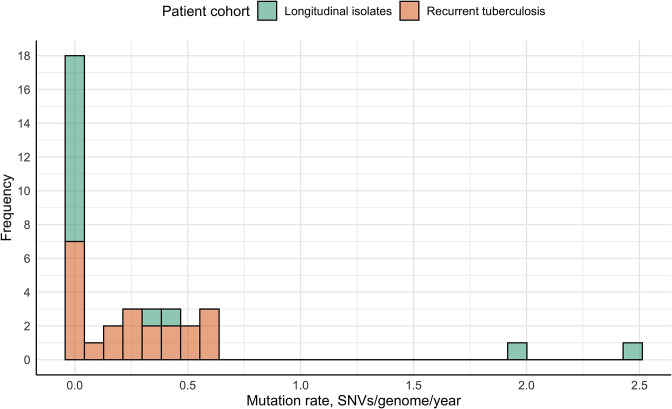
Endogenous reactivation and exogenous reinfection are two possible causes of recurrent tuberculosis (TB). However, in some cases, precise cause determination can be challenging. In this study, we used whole genome sequencing to determine pairwise SNV distances and detect differing SNVs in initial and subsequent isolates for recurrent TB cases when the first and second episodes were caused by () strains with an identical spoligotype pattern. In total, 104 isolates from 36 recurrent TB and 16 single TB episode patients were included in the study. Most isolate pairs belonged to the SIT1 (n=21), SIT42 (n=9), SIT53 (n=9), and SIT254 (n=7) spoligotypes, and in 27 cases, resistance to at least one anti-TB drug was found in either isolate. Drug susceptibility was more common in the recurrent TB patient cohort, and longitudinal single TB episode isolates were more prone to be drug-resistant (p=0.03), while the association between patient cohort and spoligotype was not statistically significant (p=0.07). The pairwise SNV-distance between the longitudinal single TB episode isolates was small (0-7 SNVs). Among the recurrent TB isolates, based on the high SNV-distance (38-273 SNVs), six reinfection cases (16.7%) were identified. This distance was small (<10 SNVs) in the remaining 30 isolate pairs. Further analysis of differing SNVs revealed that 22 (61.1%) cases could be classified as possible reactivation. Notably, despite the small distance of 2-7 SNVs, initial isolates of eight patients (22.2%) had several SNVs that were not found in the second isolates; therefore, these cases were classified as reinfection with a closely related strain. No statistically significant difference in the time interval between specimen collection in the reactivation and reinfection sample groups (p=0.13) or an association between recurrence cause and drug resistance status (p=0.62) or spoligotype (p=0.79) could be detected. The mycobacterial median mutation rate of longitudinal single TB episodes and possible reactivation isolate pairs (n=37) was 0.12 SNVs/genome/year (IQR 0-0.39), and in 18 cases (48.6%), it was equal to zero. No statistically significant differences in mutation rate were found between recurrent TB and longitudinal single TB episode isolates (p=0.087), drug-susceptible and resistant isolates (p=0.37) or isolates of Beijing and other genotype families (p=0.33). Furthermore, four cases of fluoroquinolone resistance development through the acquired SNVs in the gene were identified. To conclude, this study highlighted the complexity of recurrent episode cause determination and showed the usefulness of differing SNV identification in both isolates in such cases. Expected drug susceptibility was the only discriminative factor for recurrent TB episode-causing mycobacterial strains, while no differences between reactivation and reinfection sample groups could be identified.
内源性再激活和外源性再感染是复发性结核病(TB)的两个可能原因。然而,在某些情况下,精确的病因确定可能具有挑战性。在这项研究中,我们使用全基因组测序来确定初始和后续分离株之间的 SNV 差异,并检测复发性 TB 病例中第一次和第二次发作由具有相同 spoligotype 模式的()菌株引起时的差异 SNV。总共纳入了 36 例复发性 TB 和 16 例单发性 TB 患者的 104 株分离株。大多数分离株对属于 SIT1(n=21)、SIT42(n=9)、SIT53(n=9)和 SIT254(n=7) spoligotypes,在 27 例中,无论是在初始分离株还是在后续分离株中都发现了至少一种抗结核药物的耐药性。耐药性在复发性 TB 患者队列中更为常见,而纵向单发性 TB 发作分离株更容易耐药(p=0.03),而患者队列和 spoligotype 之间的关联没有统计学意义(p=0.07)。纵向单发性 TB 发作分离株之间的 SNV 距离较小(0-7 SNVs)。在复发性 TB 分离株中,基于高 SNV 距离(38-273 SNVs),确定了 6 例再感染病例(16.7%)。在剩余的 30 对分离株中,这一距离较小(<10 SNVs)。对差异 SNV 的进一步分析表明,22 例(61.1%)病例可归类为可能的再激活。值得注意的是,尽管 SNV 距离较小(2-7 SNVs),但 8 名患者(22.2%)的初始分离株中有几个 SNV 未在第二次分离株中发现;因此,这些病例被归类为具有密切相关菌株的再感染。在再激活和再感染样本组之间的样本采集时间间隔(p=0.13)或复发原因与耐药状态(p=0.62)或 spoligotype(p=0.79)之间未发现统计学显著差异。纵向单发性 TB 发作和可能再激活分离株对(n=37)的结核分枝杆菌中位突变率为 0.12 SNVs/基因组/年(IQR 0-0.39),在 18 例(48.6%)中,其值等于零。复发性 TB 和纵向单发性 TB 发作分离株之间的突变率无统计学显著差异(p=0.087),耐药和敏感分离株之间的突变率无统计学显著差异(p=0.37)或北京和其他基因型家族的分离株之间的突变率无统计学显著差异(p=0.33)。此外,还鉴定了 4 例通过基因中获得的 SNV 发展的氟喹诺酮类药物耐药性。总之,本研究强调了复发性发作病因确定的复杂性,并表明在这种情况下,鉴定两种分离株中的差异 SNV 是有用的。预期的药物敏感性是复发性 TB 发作相关分枝杆菌菌株的唯一鉴别因素,而无法识别再激活和再感染样本组之间的差异。