Potts David S, Torres Chris, Kwon Ohsung, Flaherty David W
Department of Chemical and Biomolecular Engineering, University of Illinois Urbana-Champaign Urbana IL 61801 USA
Chem Sci. 2023 Feb 22;14(12):3160-3181. doi: 10.1039/d2sc06473a. eCollection 2023 Mar 22.
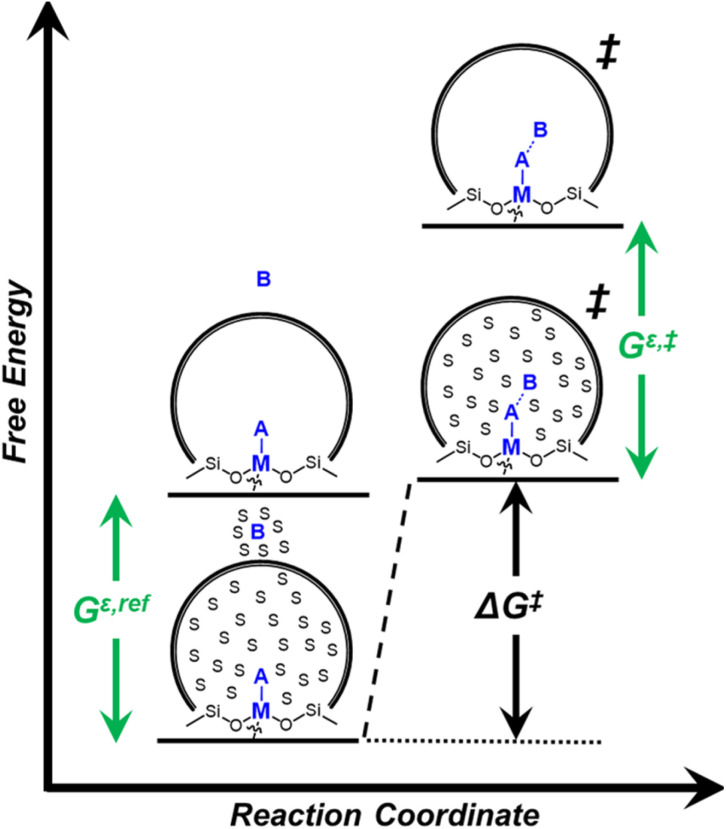
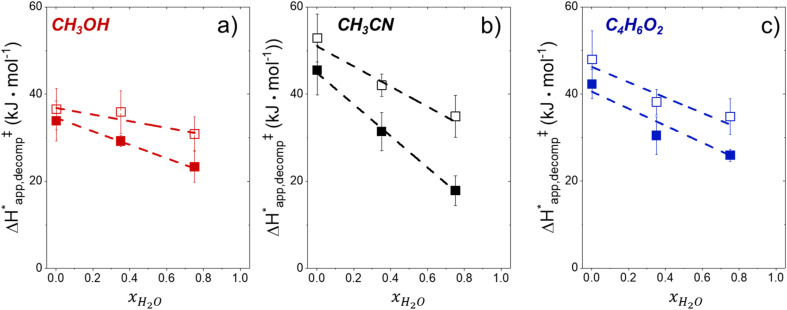
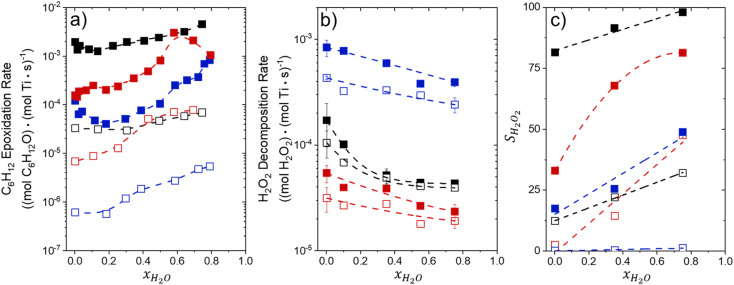
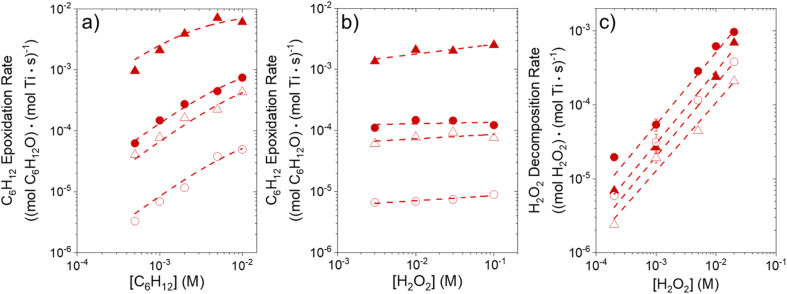
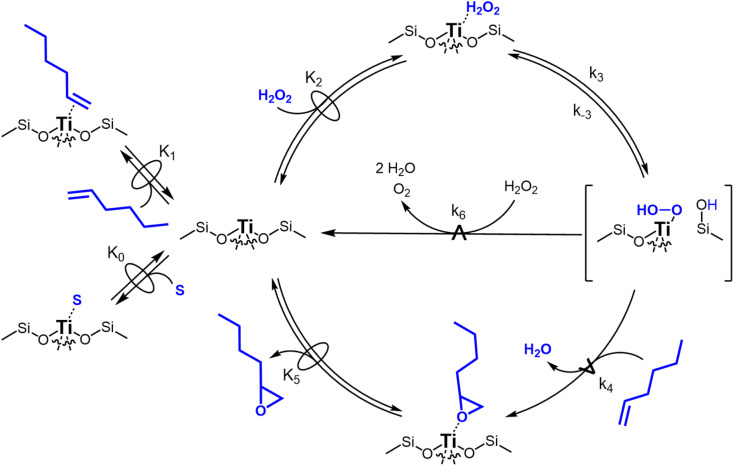
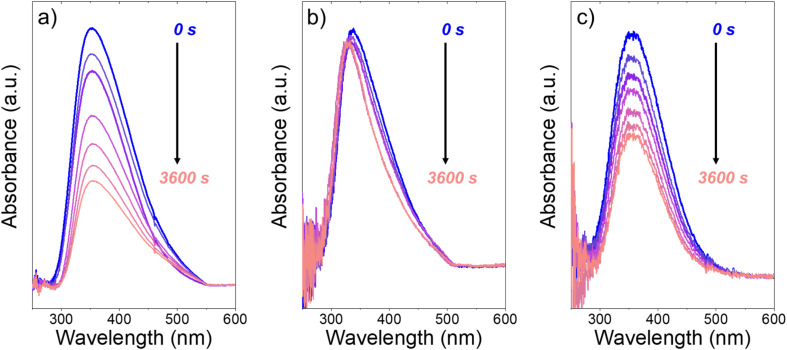
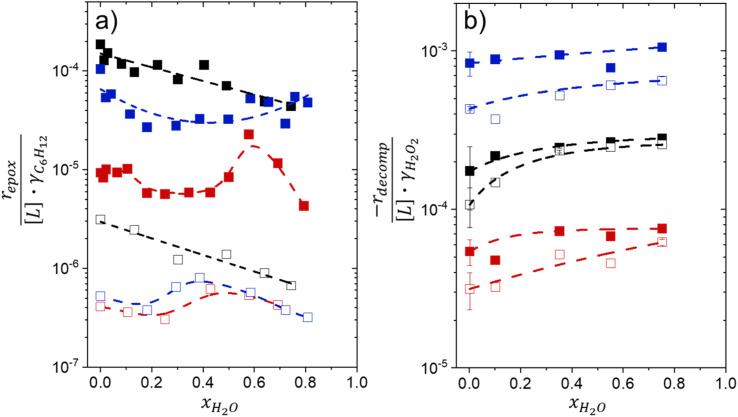
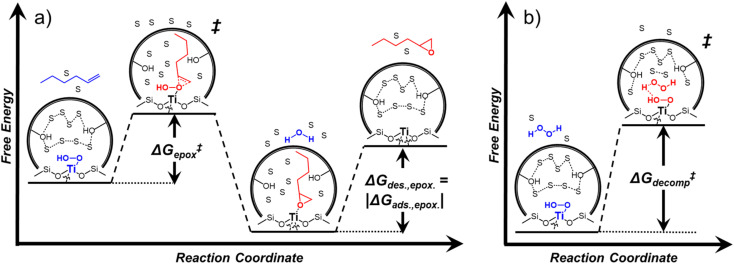
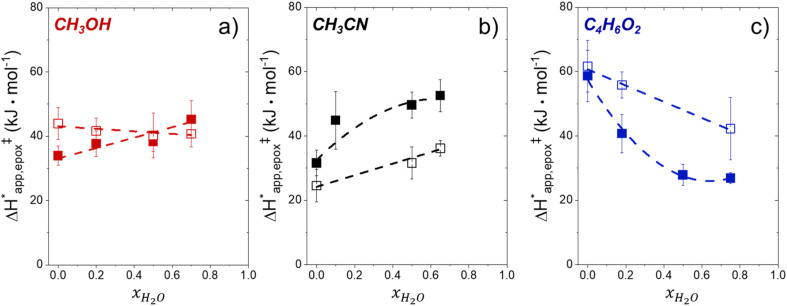
Solvent molecules alter the free energies of liquid phase species and adsorbed intermediates during catalytic reactions, thereby impacting rates and selectivities. Here, we examine these effects through the epoxidation of 1-hexene (CH) with hydrogen peroxide (HO) over hydrophilic and hydrophobic Ti-BEA zeolites immersed in aqueous solvent mixtures (acetonitrile, methanol, and γ-butyrolactone). Greater HO mole fractions provide greater epoxidation rates, lower HO decomposition rates, and hence improved HO selectivities to the desired epoxide product in each combination of solvent and zeolite. The mechanisms for epoxidation and HO decomposition remain constant across solvent compositions; however, HO activates reversibly in protic solutions. Differences in rates and selectivities reflect the disproportionate stabilization of transition states within zeolite pores with respect to surface intermediates and fluid phase reactants, as evinced by turnover rates normalized by the activity coefficients of CH and HO. Opposing trends in activation barriers suggest that the hydrophobic epoxidation transition state disrupts hydrogen bonds with solvent molecules, while the hydrophilic decomposition transition state forms hydrogen bonds with surrounding solvent molecules. Solvent compositions and adsorption volumes within pores, from H NMR spectroscopy and vapor adsorption, depend on the composition of the bulk solution and the density of silanol defects within pores. Strong correlations between epoxidation activation enthalpies and epoxide adsorption enthalpies from isothermal titration calorimetry indicate that the reorganization of solvent molecules (and associated entropy gains) required to accommodate transition states provides the most significant contribution to the stability of transition states that determine rates and selectivities. These results demonstrate that replacing a portion of organic solvents with HO offers opportunities to increase rates and selectivities for zeolite-catalyzed reactions while reducing usage of organic solvents for chemical manufacturing.
在催化反应过程中,溶剂分子会改变液相物种和吸附中间体的自由能,从而影响反应速率和选择性。在此,我们通过在浸没于含水溶剂混合物(乙腈、甲醇和γ-丁内酯)中的亲水性和疏水性钛硅沸石上用过氧化氢(H₂O₂)对1-己烯(CH)进行环氧化反应来研究这些影响。在每种溶剂和沸石的组合中,更高的H₂O₂摩尔分数可提供更高的环氧化速率、更低的H₂O₂分解速率,从而提高对所需环氧化产物的H₂O₂选择性。环氧化和H₂O₂分解的机制在不同溶剂组成下保持不变;然而,H₂O₂在质子性溶液中可逆地活化。速率和选择性的差异反映了相对于表面中间体和流体相反应物,沸石孔内过渡态的不成比例的稳定化,这通过CH和H₂O₂活度系数归一化的周转率得以证明。活化能垒的相反趋势表明,疏水性环氧化过渡态会破坏与溶剂分子的氢键,而亲水性分解过渡态会与周围溶剂分子形成氢键。通过¹H NMR光谱和蒸汽吸附得到的孔内溶剂组成和吸附体积取决于本体溶液的组成以及孔内硅醇缺陷的密度。等温滴定量热法测得的环氧化活化焓与环氧化物吸附焓之间的强相关性表明,容纳过渡态所需的溶剂分子重排(以及相关的熵增)对决定速率和选择性的过渡态稳定性贡献最大。这些结果表明,用H₂O₂替代一部分有机溶剂为提高沸石催化反应的速率和选择性提供了机会,同时减少了化学制造中有机溶剂的使用。