Institute for Medical Engineering and Science, Massachusetts Institute of Technology, Cambridge, MA 02139.
Department of Physics, Massachusetts Institute of Technology, Cambridge, MA 02139.
Proc Natl Acad Sci U S A. 2023 May 16;120(20):e2221726120. doi: 10.1073/pnas.2221726120. Epub 2023 May 8.
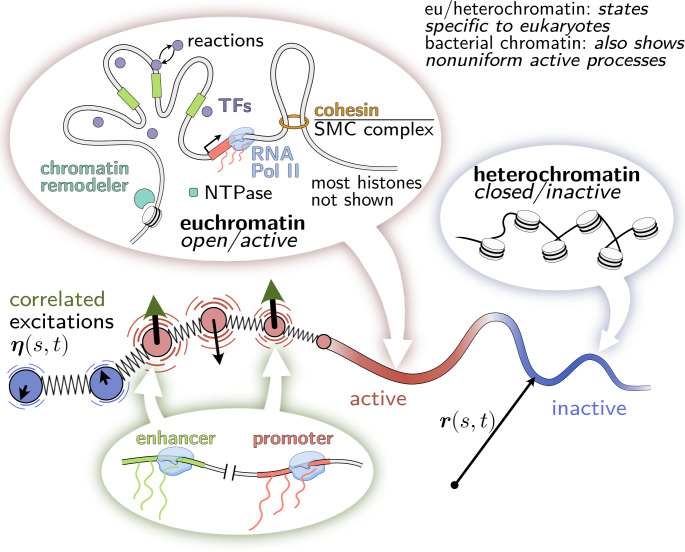
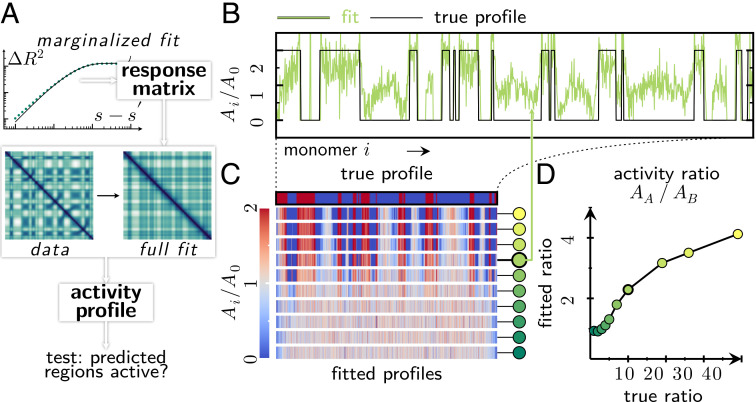
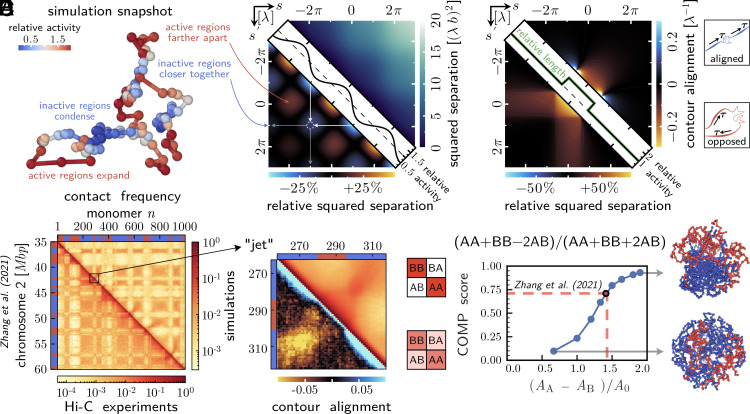
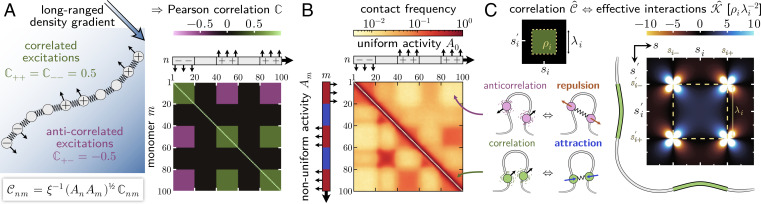
From proteins to chromosomes, polymers fold into specific conformations that control their biological function. Polymer folding has long been studied with equilibrium thermodynamics, yet intracellular organization and regulation involve energy-consuming, active processes. Signatures of activity have been measured in the context of chromatin motion, which shows spatial correlations and enhanced subdiffusion only in the presence of adenosine triphosphate. Moreover, chromatin motion varies with genomic coordinate, pointing toward a heterogeneous pattern of active processes along the sequence. How do such patterns of activity affect the conformation of a polymer such as chromatin? We address this question by combining analytical theory and simulations to study a polymer subjected to sequence-dependent correlated active forces. Our analysis shows that a local increase in activity (larger active forces) can cause the polymer backbone to bend and expand, while less active segments straighten out and condense. Our simulations further predict that modest activity differences can drive compartmentalization of the polymer consistent with the patterns observed in chromosome conformation capture experiments. Moreover, segments of the polymer that show correlated active (sub)diffusion attract each other through effective long-ranged harmonic interactions, whereas anticorrelations lead to effective repulsions. Thus, our theory offers nonequilibrium mechanisms for forming genomic compartments, which cannot be distinguished from affinity-based folding using structural data alone. As a first step toward exploring whether active mechanisms contribute to shaping genome conformations, we discuss a data-driven approach.
从蛋白质到染色体,聚合物折叠成特定的构象,控制其生物功能。聚合物折叠一直以来都是用平衡热力学来研究的,然而细胞内的组织和调节涉及到能量消耗的、主动的过程。在染色质运动的背景下,已经测量到了活性的特征,它只在三磷酸腺苷存在的情况下表现出空间相关性和增强的亚扩散。此外,染色质运动随基因组坐标而变化,这表明在序列上存在着活跃过程的异质模式。这些活性模式如何影响聚合物(如染色质)的构象?我们通过结合分析理论和模拟来研究受序列相关的主动力影响的聚合物来解决这个问题。我们的分析表明,局部活性增加(更大的主动力)会导致聚合物骨架弯曲和扩展,而活性较小的片段则变直并凝聚。我们的模拟进一步预测,适度的活性差异可以驱动聚合物的分隔化,这与染色体构象捕获实验中观察到的模式一致。此外,显示出相关主动(亚)扩散的聚合物片段通过有效的长程调和相互作用彼此吸引,而反相关则导致有效的排斥。因此,我们的理论提供了形成基因组隔室的非平衡机制,这些机制不能仅从结构数据与基于亲和力的折叠区分开来。作为探索主动机制是否有助于塑造基因组构象的第一步,我们讨论了一种数据驱动的方法。