UC Santa Cruz Genomics Institute, University of California, Santa Cruz, Santa Cruz, CA, USA.
Institute for Medical Biometry and Bioinformatics, Medical Faculty, Heinrich Heine University Düsseldorf, Düsseldorf, Germany.
Nat Biotechnol. 2024 Apr;42(4):663-673. doi: 10.1038/s41587-023-01793-w. Epub 2023 May 10.
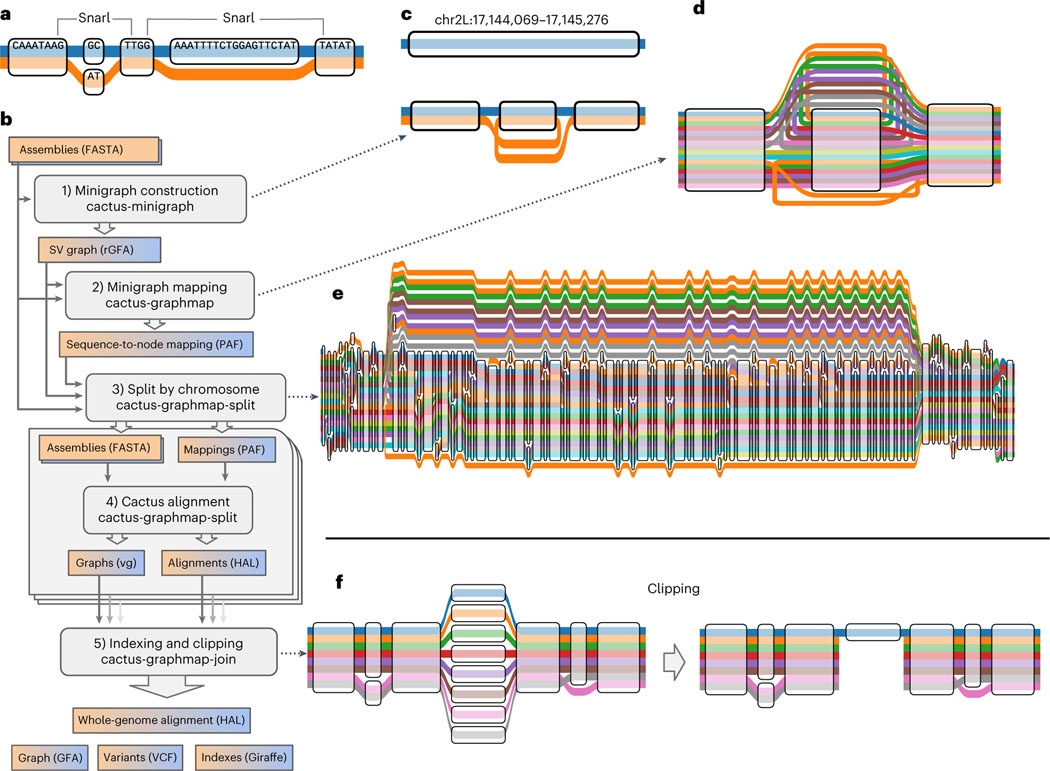
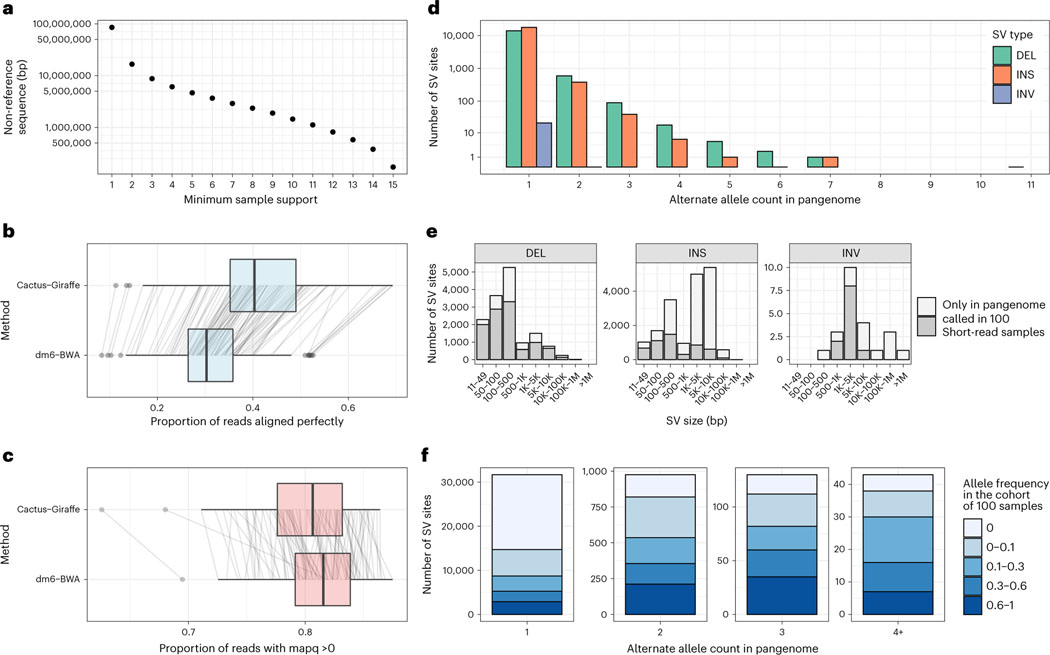
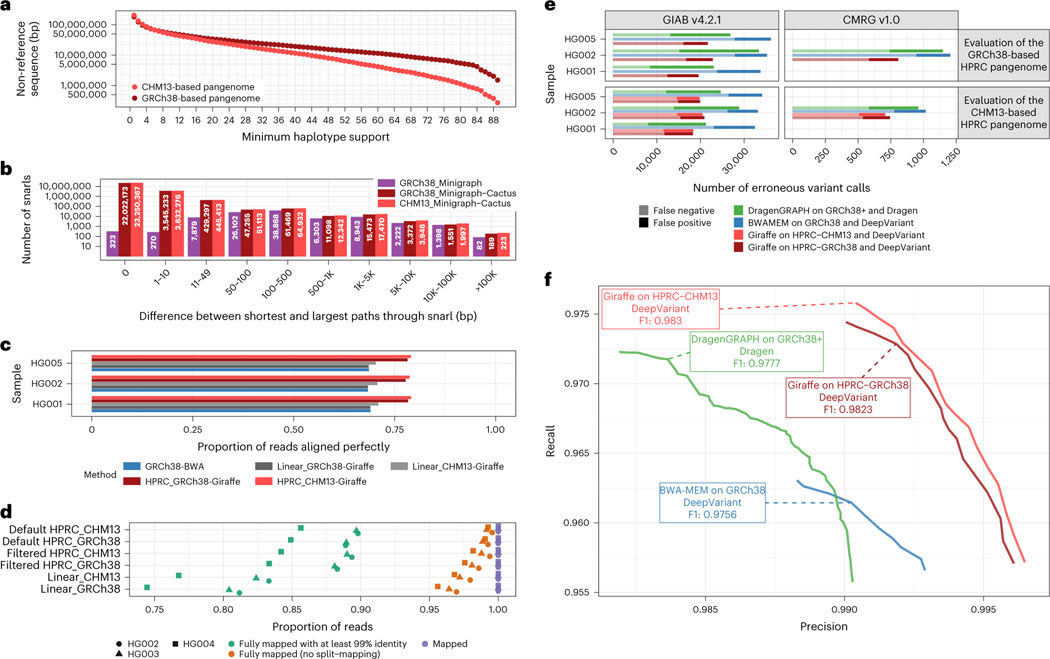
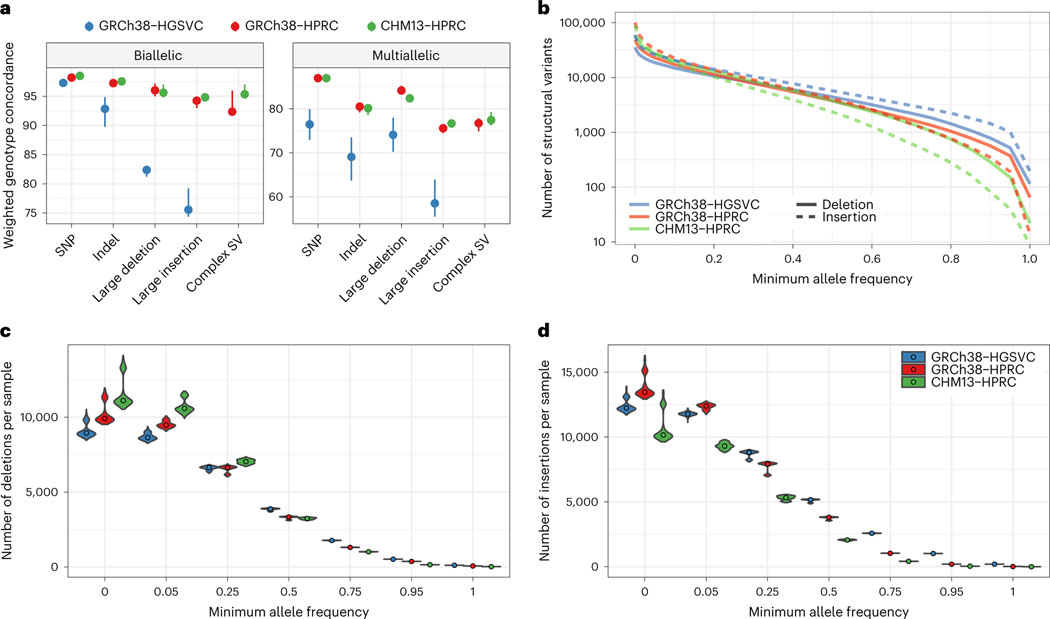
Pangenome references address biases of reference genomes by storing a representative set of diverse haplotypes and their alignment, usually as a graph. Alternate alleles determined by variant callers can be used to construct pangenome graphs, but advances in long-read sequencing are leading to widely available, high-quality phased assemblies. Constructing a pangenome graph directly from assemblies, as opposed to variant calls, leverages the graph's ability to represent variation at different scales. Here we present the Minigraph-Cactus pangenome pipeline, which creates pangenomes directly from whole-genome alignments, and demonstrate its ability to scale to 90 human haplotypes from the Human Pangenome Reference Consortium. The method builds graphs containing all forms of genetic variation while allowing use of current mapping and genotyping tools. We measure the effect of the quality and completeness of reference genomes used for analysis within the pangenomes and show that using the CHM13 reference from the Telomere-to-Telomere Consortium improves the accuracy of our methods. We also demonstrate construction of a Drosophila melanogaster pangenome.
泛基因组参考通过存储一组具有代表性的多样化单倍型及其比对,通常以图的形式,解决了参考基因组的偏差。变体调用器确定的替代等位基因可用于构建泛基因组图谱,但长读测序的进步正在导致广泛可用的高质量相组装。与从变体调用相反,直接从组装构建泛基因组图谱利用了图谱在不同尺度上表示变异的能力。在这里,我们提出了 Minigraph-Cactus 泛基因组管道,该管道直接从全基因组比对创建泛基因组,并展示了其对来自人类泛基因组参考联盟的 90 个人类单倍型的扩展能力。该方法构建了包含所有形式遗传变异的图谱,同时允许使用当前的映射和基因分型工具。我们测量了在泛基因组中用于分析的参考基因组的质量和完整性的影响,并表明使用端粒到端粒联盟的 CHM13 参考可以提高我们方法的准确性。我们还展示了黑腹果蝇泛基因组的构建。