Department of Immunology, Genetics and Pathology, Biomedical Centre, Uppsala University, Uppsala, Sweden.
Department of Clinical Genetics, and Department of Biomedical and Clinical Sciences, Linköping University, Linköping, Sweden.
Eur J Hum Genet. 2024 Mar;32(3):333-341. doi: 10.1038/s41431-023-01392-y. Epub 2023 Jun 5.
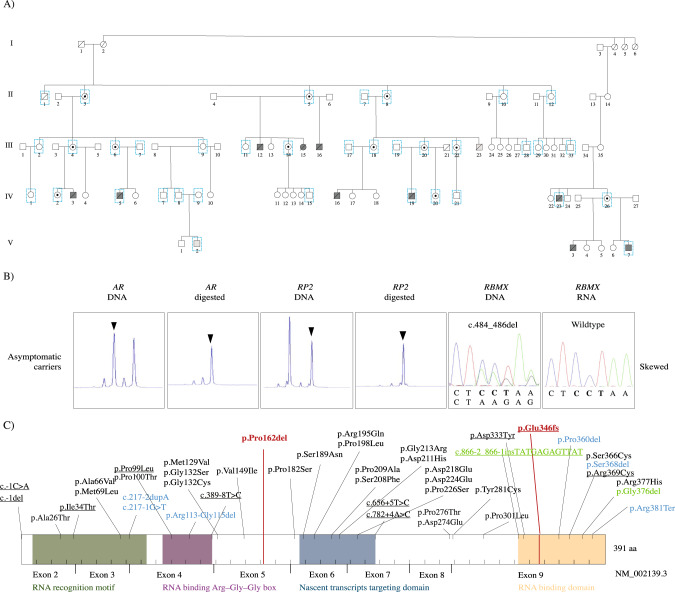
RNA binding motif protein X-linked (RBMX) encodes the heterogeneous nuclear ribonucleoprotein G (hnRNP G) that regulates splicing, sister chromatid cohesion and genome stability. RBMX knock down experiments in various model organisms highlight the gene's importance for brain development. Deletion of the RGG/RG motif in hnRNP G has previously been associated with Shashi syndrome, however involvement of other hnRNP G domains in intellectual disability remain unknown. In the current study, we present the underlying genetic and molecular cause of Gustavson syndrome. Gustavson syndrome was first reported in 1993 in a large Swedish five-generation family presented with profound X-linked intellectual disability and an early death. Extensive genomic analyses of the family revealed hemizygosity for a novel in-frame deletion in RBMX in affected individuals (NM_002139.4; c.484_486del, p.(Pro162del)). Carrier females were asymptomatic and presented with skewed X-chromosome inactivation, indicating silencing of the pathogenic allele. Affected individuals presented minor phenotypic overlap with Shashi syndrome, indicating a different disease-causing mechanism. Investigation of the variant effect in a neuronal cell line (SH-SY5Y) revealed differentially expressed genes enriched for transcription factors involved in RNA polymerase II transcription. Prediction tools and a fluorescence polarization assay imply a novel SH3-binding motif of hnRNP G, and potentially a reduced affinity to SH3 domains caused by the deletion. In conclusion, we present a novel in-frame deletion in RBMX segregating with Gustavson syndrome, leading to disturbed RNA polymerase II transcription, and potentially reduced SH3 binding. The results indicate that disruption of different protein domains affects the severity of RBMX-associated intellectual disabilities.
X 连锁 RNA 结合基序蛋白(RBMX)编码异质核核糖核蛋白 G(hnRNP G),该蛋白调节剪接、姐妹染色单体黏合和基因组稳定性。在各种模式生物中进行的 RBMX 敲低实验突出了该基因对大脑发育的重要性。hnRNP G 中的 RGG/RG 基序缺失以前与 Shashi 综合征有关,但是其他 hnRNP G 结构域在智力障碍中的参与仍然未知。在本研究中,我们介绍了 Gustavson 综合征的潜在遗传和分子原因。Gustavson 综合征于 1993 年在一个大型瑞典五代家系中首次报道,该家系表现为严重的 X 连锁智力障碍和早逝。对该家系的广泛基因组分析显示,受影响个体存在 RBMX 中新型框内缺失的杂合性(NM_002139.4;c.484_486del,p.(Pro162del))。携带女性无症状且表现出偏性 X 染色体失活,表明致病等位基因沉默。受影响的个体与 Shashi 综合征表现出轻微的表型重叠,表明存在不同的致病机制。在神经元细胞系(SH-SY5Y)中对变体效应的研究表明,差异表达的基因富集了参与 RNA 聚合酶 II 转录的转录因子。预测工具和荧光偏振测定法暗示 hnRNP G 的新型 SH3 结合基序,并且由于缺失导致与 SH3 结构域的结合亲和力降低。总之,我们提出了一种与 Gustavson 综合征分离的 RBMX 中的新型框内缺失,导致 RNA 聚合酶 II 转录紊乱,并且潜在地降低了 SH3 结合。结果表明,不同蛋白结构域的破坏会影响 RBMX 相关智力障碍的严重程度。