Bioinformatics, Biotechnology Center (BIOTEC), Technische Universität Dresden, Dresden, Saxony, Germany.
University of Verona, Verona, Italy.
Sci Rep. 2023 Jun 6;13(1):9204. doi: 10.1038/s41598-023-35671-x.
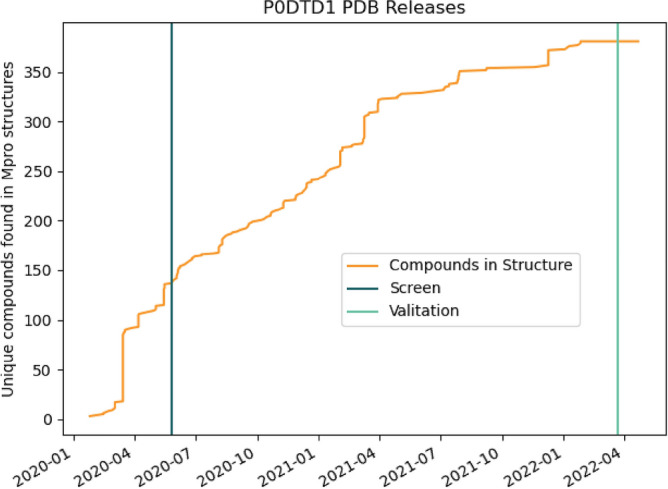
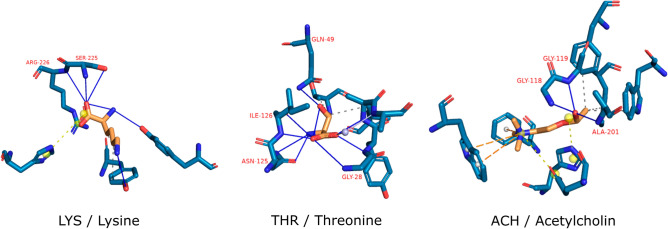
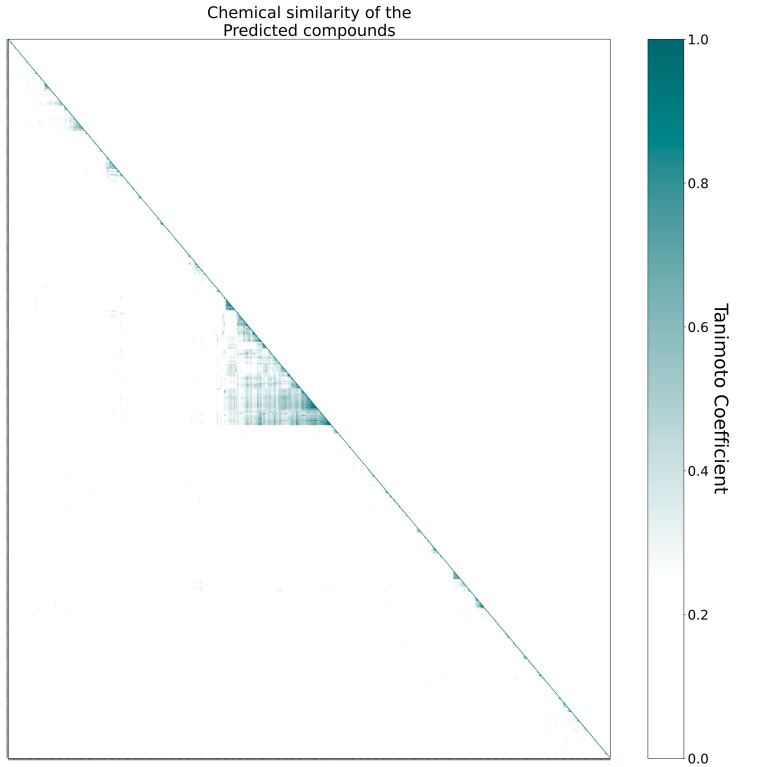
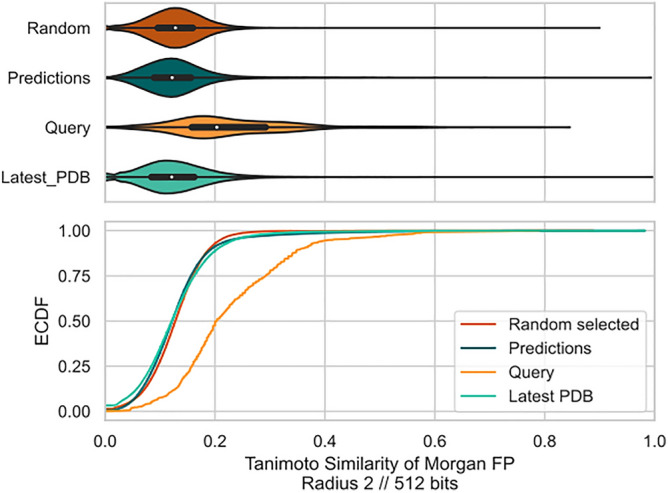
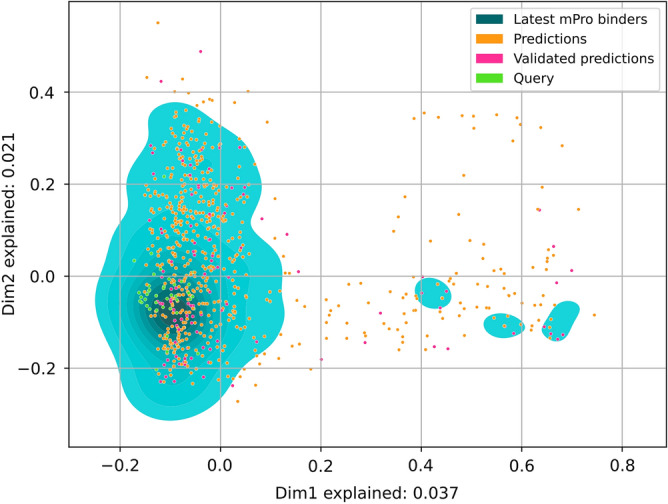
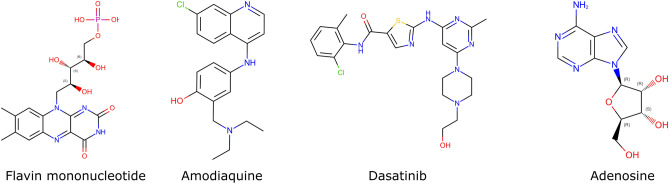
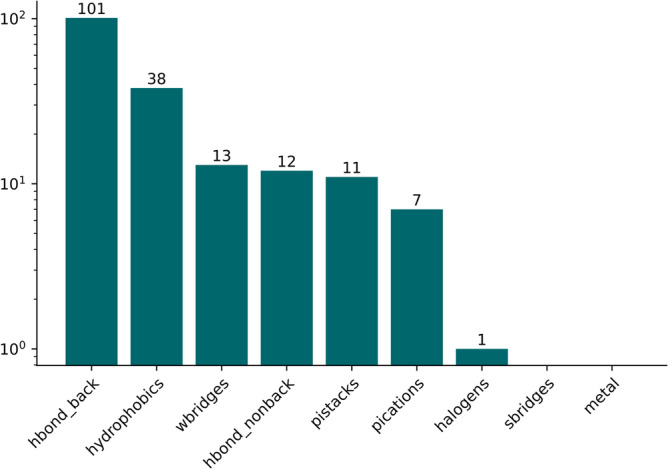
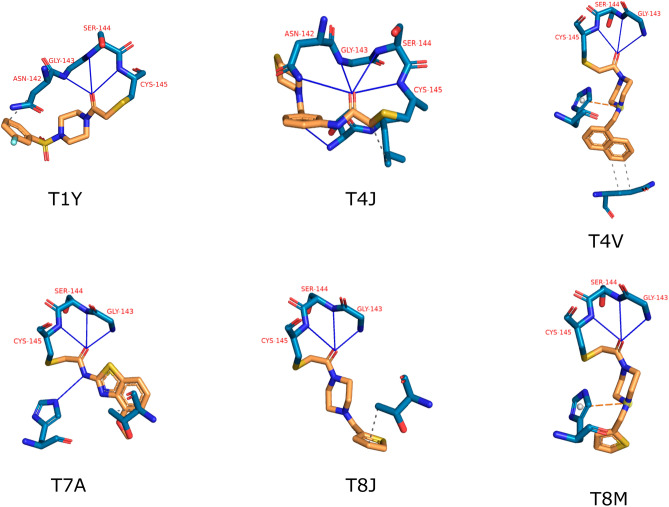
The recent outbreak of the COVID-19 pandemic caused by severe acute respiratory syndrome-Coronavirus-2 (SARS-CoV-2) has shown the necessity for fast and broad drug discovery methods to enable us to react quickly to novel and highly infectious diseases. A well-known SARS-CoV-2 target is the viral main 3-chymotrypsin-like cysteine protease (M), known to control coronavirus replication, which is essential for the viral life cycle. Here, we applied an interaction-based drug repositioning algorithm on all protein-compound complexes available in the protein database (PDB) to identify M inhibitors and potential novel compound scaffolds against SARS-CoV-2. The screen revealed a heterogeneous set of 692 potential M inhibitors containing known ones such as Dasatinib, Amodiaquine, and Flavin mononucleotide, as well as so far untested chemical scaffolds. In a follow-up evaluation, we used publicly available data published almost two years after the screen to validate our results. In total, we are able to validate 17% of the top 100 predictions with publicly available data and can furthermore show that predicted compounds do cover scaffolds that are yet not associated with M. Finally, we detected a potentially important binding pattern consisting of 3 hydrogen bonds with hydrogen donors of an oxyanion hole within the active side of M. Overall, these results give hope that we will be better prepared for future pandemics and that drug development will become more efficient in the upcoming years.
近期由严重急性呼吸系统综合症冠状病毒 2 型(SARS-CoV-2)引发的 COVID-19 疫情爆发,凸显了我们需要快速广泛地应用药物发现方法,以便能够对新型和高传染性疾病迅速做出反应。众所周知,SARS-CoV-2 的一个靶标是病毒的主要 3-糜蛋白酶样半胱氨酸蛋白酶(M),该蛋白酶已知可控制冠状病毒复制,这对病毒的生命周期至关重要。在这里,我们将基于相互作用的药物重定位算法应用于蛋白质数据库(PDB)中所有可用的蛋白质-化合物复合物,以鉴定 M 抑制剂和针对 SARS-CoV-2 的潜在新型化合物骨架。筛选结果揭示了一组包含达沙替尼、阿莫地喹和黄素单核苷酸等已知抑制剂以及迄今未经测试的化学骨架的 692 种潜在 M 抑制剂。在后续评估中,我们使用了在筛选后将近两年公布的公开可用数据来验证我们的结果。总的来说,我们能够用公开可用的数据验证前 100 名预测中的 17%,并且还可以证明预测的化合物确实涵盖了尚未与 M 相关联的骨架。最后,我们检测到一个潜在的重要结合模式,该模式由 3 个氢键组成,与 M 的活性部位中的一个阴离子空穴的氢供体形成氢键。总体而言,这些结果表明我们将为未来的大流行做好更好的准备,并且在未来几年药物开发将变得更加高效。