Castellanos Maria A, Payne Alexander M, Scheen Jenke, MacDermott-Opeskin Hugo, Pulido Iván, Balcomb Blake H, Griffen Ed J, Fearon Daren, Barr Haim, Lahav Noa, Cousins David, Stacey Jessica, Robinson Ralph, Lefker Bruce, Chodera John D
Computational and Systems Biology Program, Memorial Sloan Kettering Cancer Center, New York, NY, USA.
Tri-Institutional Ph.D. Program in Chemical Biology, Weill Cornell Medical College of Cornell University, New York, NY, USA.
bioRxiv. 2025 Jul 30:2025.07.29.667267. doi: 10.1101/2025.07.29.667267.
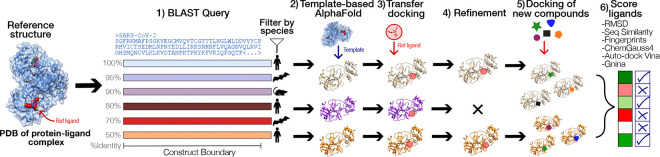
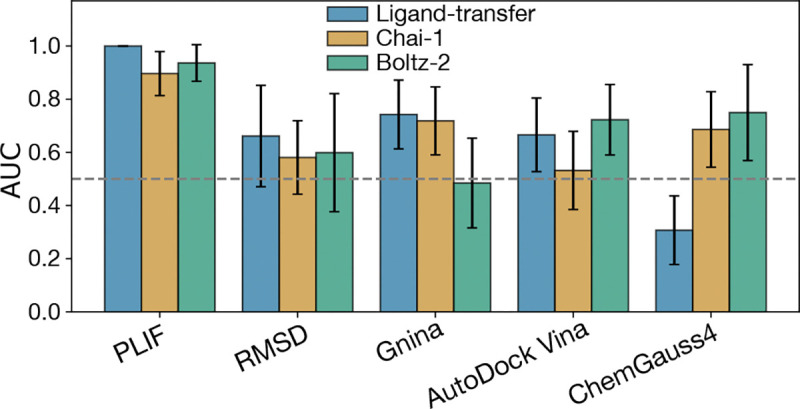
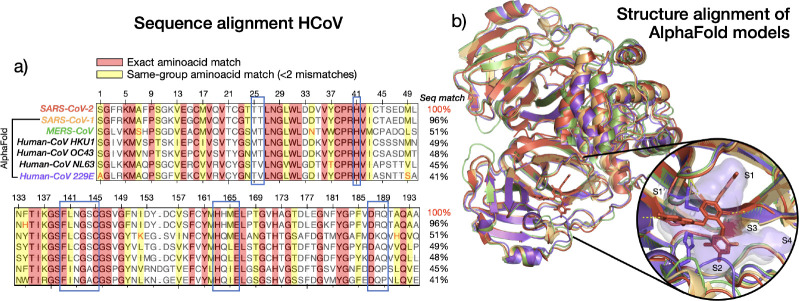
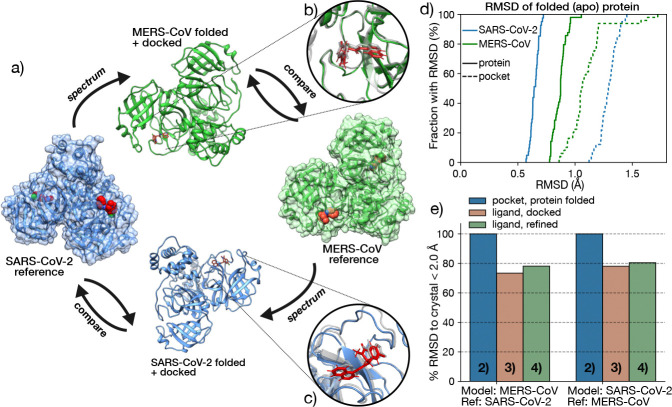
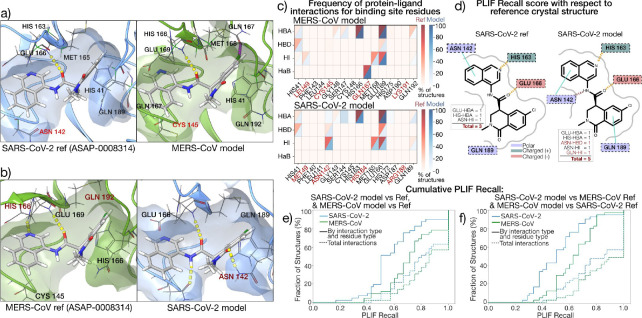
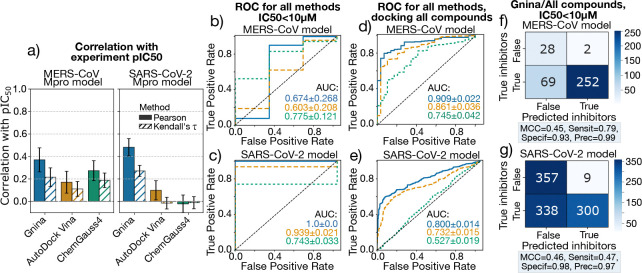
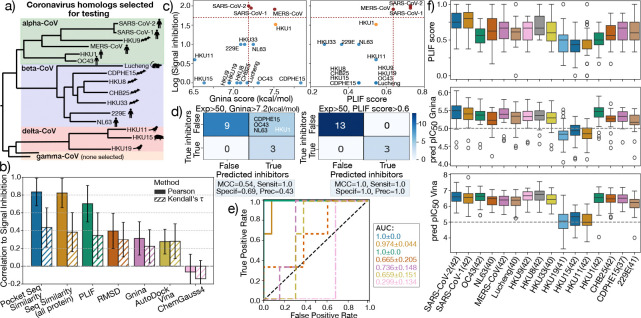
The rapid emergence of viruses with pandemic potential continues to pose a threat to public health worldwide. With the typical drug discovery pipeline taking an average of 5-10 years to reach clinical readiness, there is an urgent need for strategies to develop broad-spectrum antivirals that can target multiple viral family members and variants of concern. We present a structure-based computational pipeline designed to identify and evaluate broad-spectrum inhibitors across viral family members for a given target in order to support spectrum breadth assessment and prioritization in lead optimization programs. This pipeline comprises three key steps: (1) an automated search to identify viral sequences related to a specified target construct, (2) pose prediction leveraging any available structural data, and (3) scoring of protein-ligand complexes to estimate antiviral activity breadth. The pipeline is implemented using the drugforge package: an open-source toolkit for structure-based antiviral discovery. To validate this framework, we retrospectively evaluated two overlapping datasets of ligands bound to the SARS-CoV-2 and MERS-CoV main protease (M), observing useful predictive power with respect to experimental binding affinities. Additionally, we screened known SARS-CoV-2 M inhibitors against a panel of human and non-human coronaviruses, demonstrating the potential of this approach to assess broad-spectrum antiviral activity. Our computational strategy aims to accelerate the identification of antiviral therapies for current and emerging viruses with pandemic potential, contributing to global preparedness for future outbreaks.
具有大流行潜力的病毒迅速出现,继续对全球公共卫生构成威胁。由于典型的药物研发流程平均需要5到10年才能达到临床应用准备阶段,因此迫切需要开发能够针对多个病毒家族成员和相关变体的广谱抗病毒药物的策略。我们提出了一种基于结构的计算流程,旨在识别和评估针对给定靶点的跨病毒家族成员的广谱抑制剂,以支持先导优化计划中的活性谱广度评估和优先级排序。该流程包括三个关键步骤:(1)自动搜索以识别与指定靶点构建体相关的病毒序列,(2)利用任何可用的结构数据进行构象预测,以及(3)对蛋白质-配体复合物进行评分以估计抗病毒活性广度。该流程使用drugforge软件包实现:一个用于基于结构的抗病毒药物发现的开源工具包。为了验证这个框架,我们回顾性评估了与SARS-CoV-2和MERS-CoV主要蛋白酶(M)结合的两个重叠配体数据集,观察到相对于实验结合亲和力具有有用的预测能力。此外,我们针对一组人类和非人类冠状病毒筛选了已知的SARS-CoV-2 M抑制剂,证明了这种方法评估广谱抗病毒活性的潜力。我们的计算策略旨在加速识别针对当前和具有大流行潜力的新兴病毒的抗病毒疗法,为全球应对未来疫情做好准备。