Neuro-SysMed Center of Excellence, Department of Neurology, Department of Clinical Medicine, Haukeland University Hospital, University of Bergen, 5021, Bergen, Norway.
Department of Clinical Medicine, University of Bergen, Pb 7804, 5020, Bergen, Norway.
Genome Med. 2023 Jun 7;15(1):41. doi: 10.1186/s13073-023-01195-2.
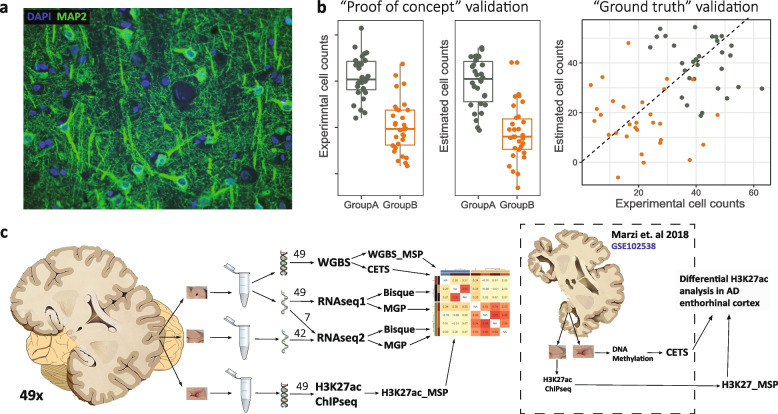
Variation in cell composition can dramatically impact analyses in bulk tissue samples. A commonly employed approach to mitigate this issue is to adjust statistical models using estimates of cell abundance derived directly from omics data. While an arsenal of estimation methods exists, the applicability of these methods to brain tissue data and whether or not cell estimates can sufficiently account for confounding cellular composition has not been adequately assessed.
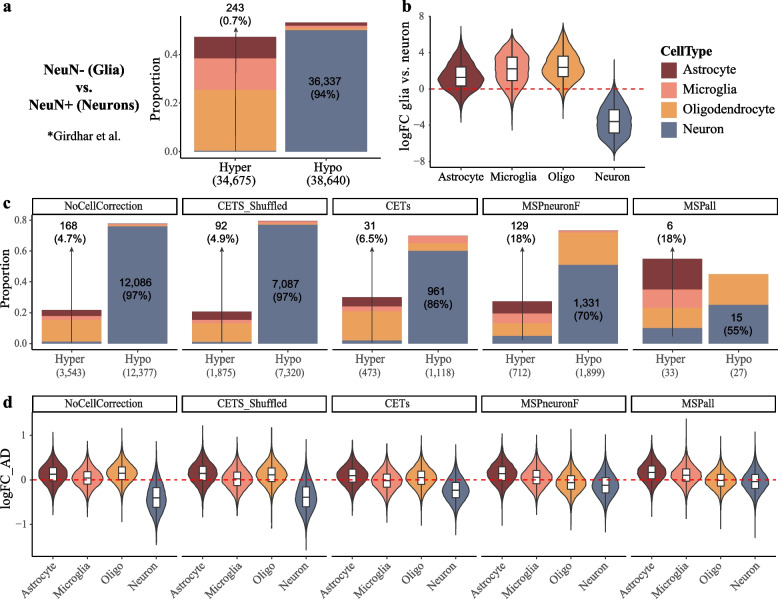
We assessed the correspondence between different estimation methods based on transcriptomic (RNA sequencing, RNA-seq) and epigenomic (DNA methylation and histone acetylation) data from brain tissue samples of 49 individuals. We further evaluated the impact of different estimation approaches on the analysis of H3K27 acetylation chromatin immunoprecipitation sequencing (ChIP-seq) data from entorhinal cortex of individuals with Alzheimer's disease and controls.
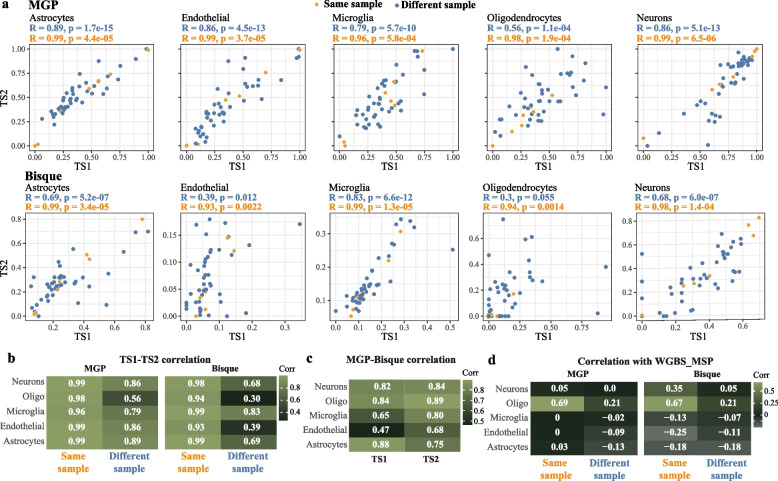
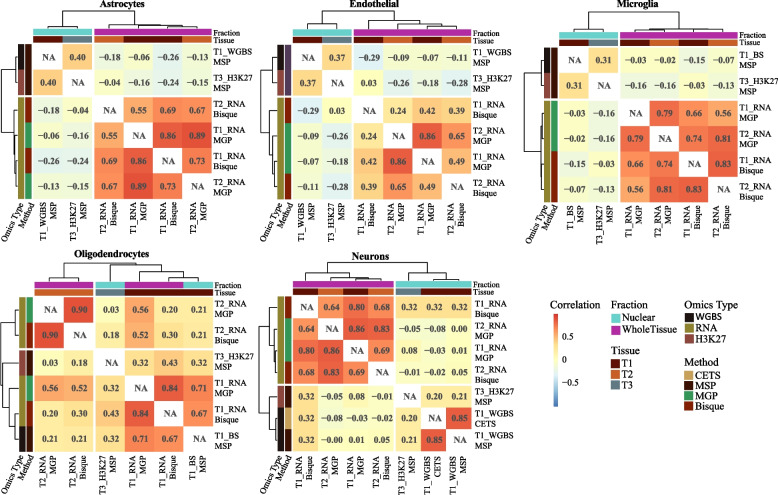
We show that even closely adjacent tissue samples from the same Brodmann area vary greatly in their cell composition. Comparison across different estimation methods indicates that while different estimation methods applied to the same data produce highly similar outcomes, there is a surprisingly low concordance between estimates based on different omics data modalities. Alarmingly, we show that cell type estimates may not always sufficiently account for confounding variation in cell composition.
Our work indicates that cell composition estimation or direct quantification in one tissue sample should not be used as a proxy to the cellular composition of another tissue sample from the same brain region of an individual-even if the samples are directly adjacent. The highly similar outcomes observed among vastly different estimation methods, highlight the need for brain benchmark datasets and better validation approaches. Finally, unless validated through complementary experiments, the interpretation of analyses outcomes based on data confounded by cell composition should be done with great caution, and ideally avoided all together.
细胞组成的差异会极大地影响批量组织样本的分析。一种常用的方法是使用直接从组学数据中得出的细胞丰度估计值来调整统计模型。虽然存在许多估计方法,但这些方法是否适用于脑组织数据,以及细胞估计值是否足以解释细胞组成的混杂问题,尚未得到充分评估。
我们评估了基于 49 个人脑组织样本的转录组(RNA 测序,RNA-seq)和表观基因组(DNA 甲基化和组蛋白乙酰化)数据的不同估计方法之间的对应关系。我们进一步评估了不同估计方法对阿尔茨海默病患者和对照者额皮质 H3K27 乙酰化染色质免疫沉淀测序(ChIP-seq)数据分析的影响。
我们表明,即使是来自同一布罗德曼区的紧密相邻的组织样本,其细胞组成也存在很大差异。不同估计方法的比较表明,尽管应用于同一数据的不同估计方法产生了高度相似的结果,但基于不同组学数据模式的估计之间的一致性却出奇地低。令人震惊的是,我们表明细胞类型估计值并不总是足以解释细胞组成的混杂变化。
我们的工作表明,即使是来自同一大脑区域的紧密相邻的组织样本,也不应该将一个组织样本的细胞组成估计值或直接定量值作为另一个组织样本的细胞组成的替代值。虽然观察到的不同估计方法之间的高度相似结果,但需要更好的基准数据集和验证方法。最后,除非通过补充实验进行验证,否则基于受细胞组成混杂影响的数据的分析结果的解释应该非常谨慎,如果可能的话,最好完全避免。