School of Biotechnology and Biomolecular Sciences, University of New South Wales, Sydney, NSW, Australia.
Harry Perkins Institute of Medical Research, QEII Medical Centre and Centre for Medical Research, The University of Western Australia, Perth, WA, Australia.
Nat Commun. 2022 Mar 15;13(1):1358. doi: 10.1038/s41467-022-28655-4.
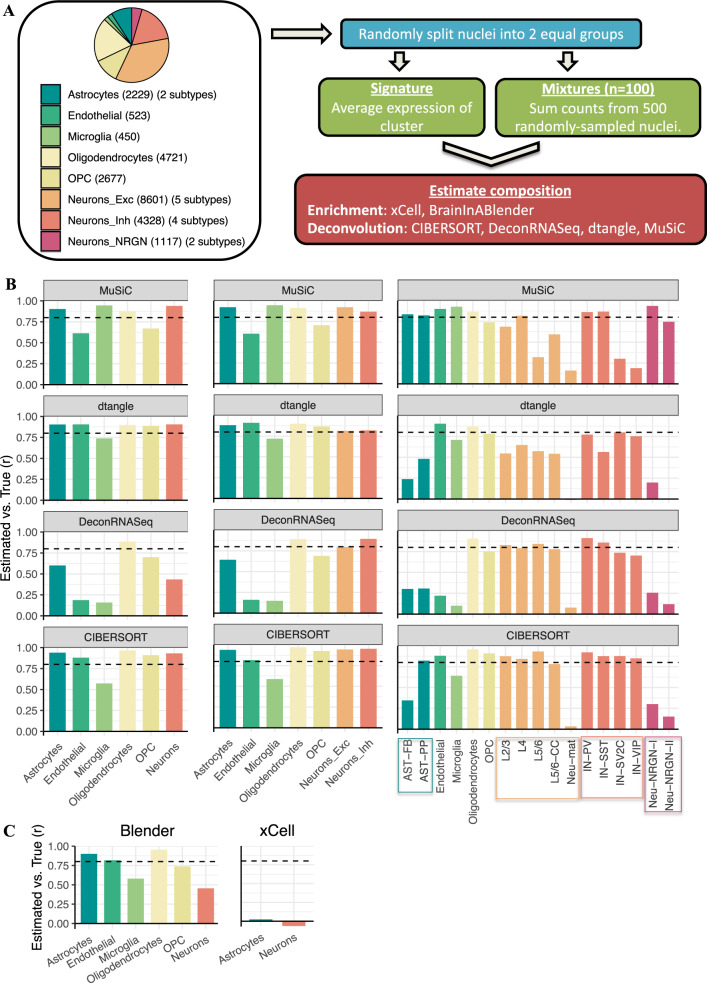
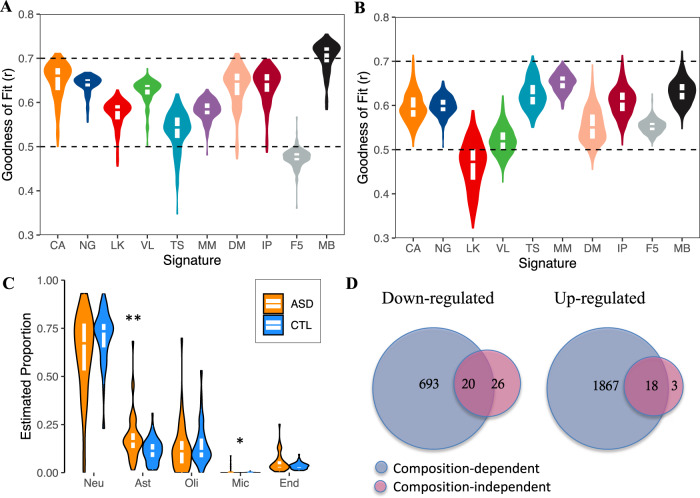
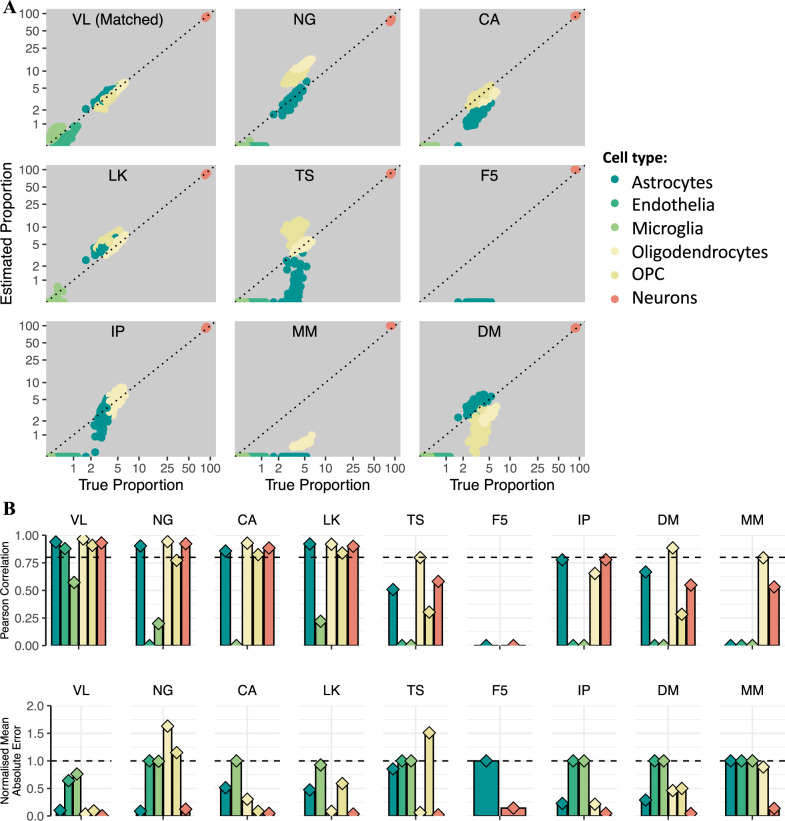
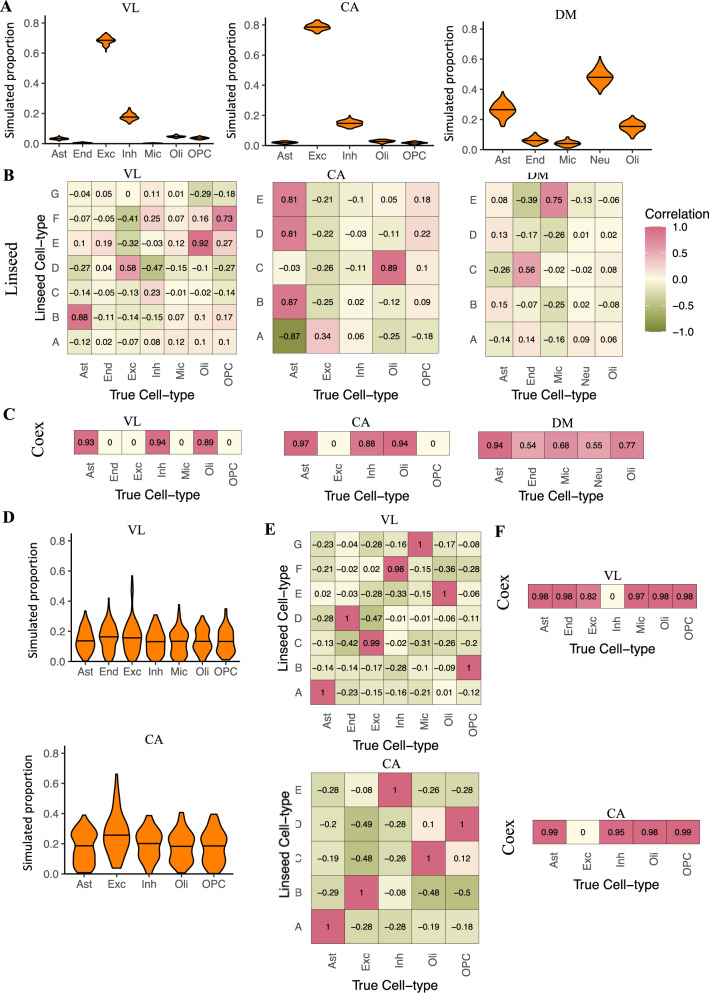
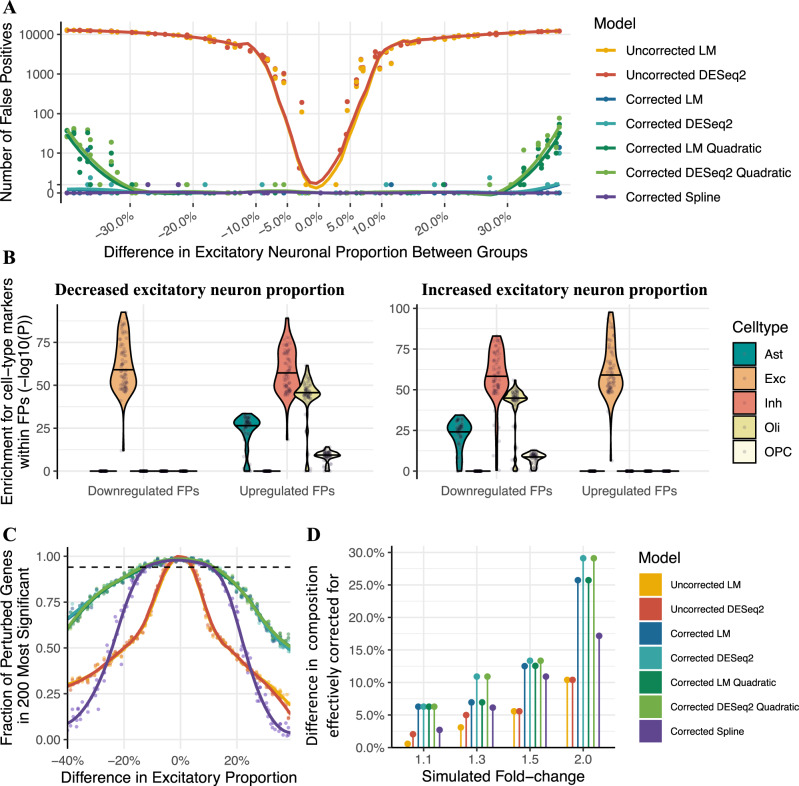
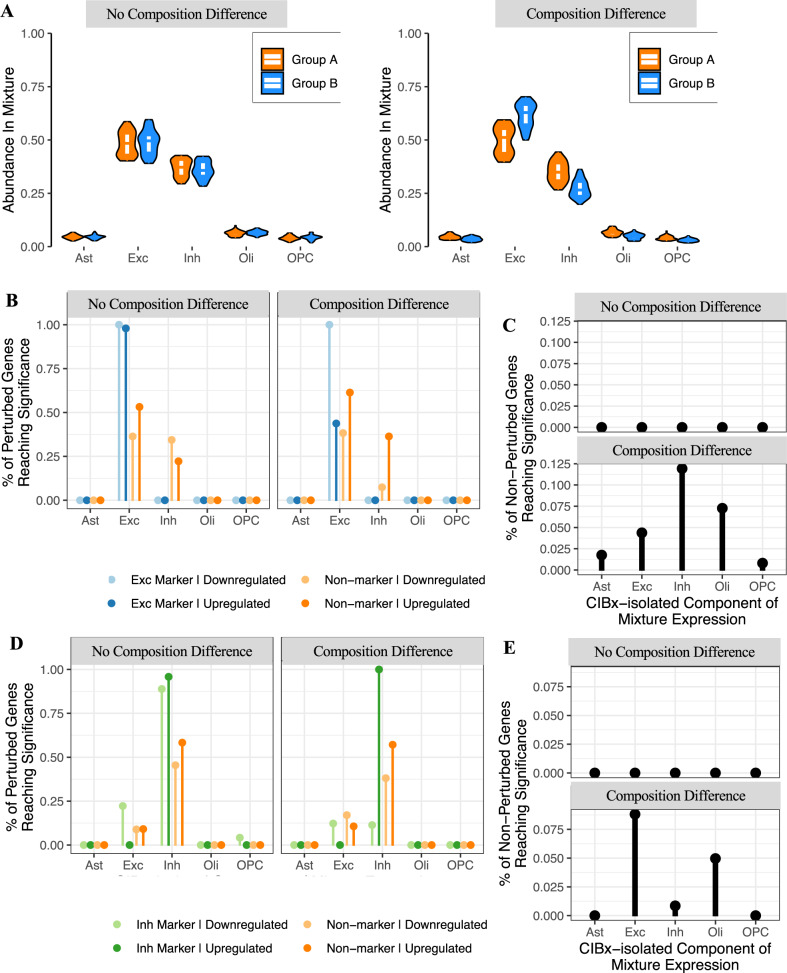
Transcriptome deconvolution aims to estimate the cellular composition of an RNA sample from its gene expression data, which in turn can be used to correct for composition differences across samples. The human brain is unique in its transcriptomic diversity, and comprises a complex mixture of cell-types, including transcriptionally similar subtypes of neurons. Here, we carry out a comprehensive evaluation of deconvolution methods for human brain transcriptome data, and assess the tissue-specificity of our key observations by comparison with human pancreas and heart. We evaluate eight transcriptome deconvolution approaches and nine cell-type signatures, testing the accuracy of deconvolution using in silico mixtures of single-cell RNA-seq data, RNA mixtures, as well as nearly 2000 human brain samples. Our results identify the main factors that drive deconvolution accuracy for brain data, and highlight the importance of biological factors influencing cell-type signatures, such as brain region and in vitro cell culturing.
转录组去卷积旨在根据基因表达数据估计 RNA 样本的细胞组成,而这反过来又可用于校正样本之间的组成差异。人脑在转录组多样性方面具有独特性,由多种细胞类型组成,包括转录相似的神经元亚型。在这里,我们对人类大脑转录组数据的去卷积方法进行了全面评估,并通过与人类胰腺和心脏的比较来评估我们的关键观察结果的组织特异性。我们评估了八种转录组去卷积方法和九种细胞类型特征,使用单细胞 RNA-seq 数据、RNA 混合物以及近 2000 个人脑样本的计算机模拟混合物来测试去卷积的准确性。我们的结果确定了驱动大脑数据去卷积准确性的主要因素,并强调了影响细胞类型特征的生物学因素的重要性,例如大脑区域和体外细胞培养。