Di Salvatore Valentina, Crispino Elena, Maleki Avisa, Nicotra Giulia, Russo Giulia, Pappalardo Francesco
Department of Drug and Health Sciences, University of Catania, Catania, Italy.
Department of Biomedical and Biotechnological Sciences, University of Catania, Catania, Italy.
Comput Struct Biotechnol J. 2023;21:3339-3354. doi: 10.1016/j.csbj.2023.06.007. Epub 2023 Jun 12.
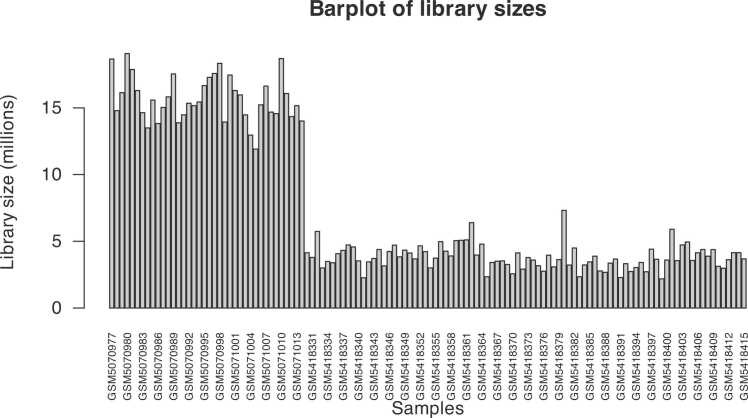
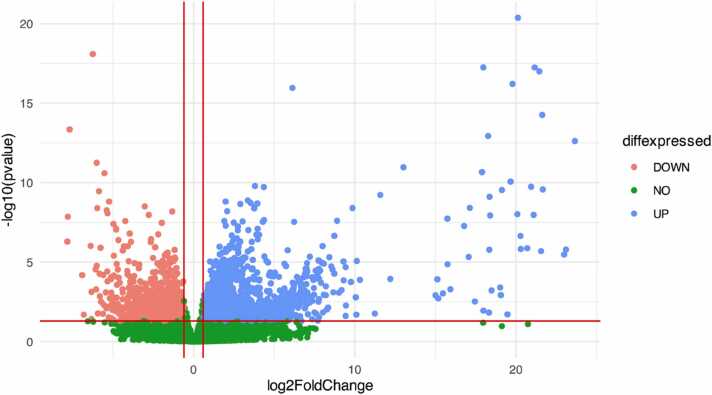
COVID-19 was declared a pandemic in March 2020, and since then, it has not stopped spreading like wildfire in almost every corner of the world, despite the many efforts made to stem its spread. SARS-CoV-2 has one of the biggest genomes among RNA viruses and presents unique characteristics that differentiate it from other coronaviruses, making it even more challenging to find a cure or vaccine that is efficient enough. This work aims, using RNA sequencing (RNA-Seq) data, to evaluate whether the expression of specific human genes in the host can vary in different grades of disease severity and to determine the molecular origins of the differences in response to SARS-CoV-2 infection in different patients. In addition to quantifying gene expression, data coming from RNA-Seq allow for the discovery of new transcripts, the identification of alternative splicing events, the detection of allele-specific expression, and the detection of post-transcriptional alterations. For this reason, we performed differential expression analysis on different expression profiles of COVID-19 patients, using RNA-Seq data coming from NCBI public repository, and we obtained the lists of all differentially expressed genes (DEGs) emerging from 7 experimental conditions. We performed a Gene Set Enrichment Analysis (GSEA) on these genes to find possible correlations between DEGs and known disease phenotypes. We mainly focused on DEGs coming out from the analysis of the contrasts involving severe conditions to infer any possible relation between a worsening of the clinical picture and an over-representation of specific genes. Based on the obtained results, this study indicates a small group of genes that result up-regulated in the severe form of the disease. EXOSC5, MESD, REXO2, and TRMT2A genes are not differentially expressed or not present in the other conditions, being for that reason, good biomarkers candidates for the severe form of COVID-19 disease. The use of specific over-expressed genes, whether up-regulated or down-regulated, which have an individual role in each different condition of COVID-19 as a biomarker, can assist in early diagnosis.
2020年3月,新型冠状病毒肺炎(COVID-19)被宣布为大流行病,自那时以来,尽管人们为阻止其传播付出了诸多努力,但它仍在世界几乎每个角落如野火般蔓延。严重急性呼吸综合征冠状病毒2(SARS-CoV-2)拥有RNA病毒中最大的基因组之一,并呈现出使其有别于其他冠状病毒的独特特征,这使得找到一种足够有效的治疗方法或疫苗更具挑战性。这项工作旨在利用RNA测序(RNA-Seq)数据,评估宿主中特定人类基因的表达在不同疾病严重程度等级下是否会有所不同,并确定不同患者对SARS-CoV-2感染反应差异的分子根源。除了定量基因表达外,RNA-Seq数据还能用于发现新转录本、识别可变剪接事件、检测等位基因特异性表达以及检测转录后改变。因此,我们使用来自美国国立医学图书馆(NCBI)公共数据库的RNA-Seq数据,对COVID-19患者的不同表达谱进行差异表达分析,并获得了7种实验条件下所有差异表达基因(DEG)的列表。我们对这些基因进行了基因集富集分析(GSEA),以发现DEG与已知疾病表型之间的可能关联。我们主要关注从涉及重症情况的对比分析中得出的DEG,以推断临床症状恶化与特定基因过度表达之间的任何可能关系。基于所得结果,本研究表明一小部分基因在疾病的重症形式中上调。外切体复合物亚基5(EXOSC5)、mesoderm development organizer(MESD)、核糖核酸外切酶2(REXO2)和甲基转移酶2A(TRMT2A)基因在其他条件下无差异表达或不存在,因此是COVID-19重症形式的良好生物标志物候选基因。使用在COVID-19的每种不同情况下具有个体作用的特定过表达基因(无论是上调还是下调)作为生物标志物,有助于早期诊断。