Department of Computer Science, Johns Hopkins University, Baltimore, MD, USA.
Department of Biochemistry and Molecular Biology, Penn State University, College Park, PA, USA.
BMC Bioinformatics. 2023 Jun 23;24(1):263. doi: 10.1186/s12859-023-05389-8.
Protein-protein interactions play a crucial role in almost all cellular processes. Identifying interacting proteins reveals insight into living organisms and yields novel drug targets for disease treatment. Here, we present a publicly available, automated pipeline to predict genome-wide protein-protein interactions and produce high-quality multimeric structural models.
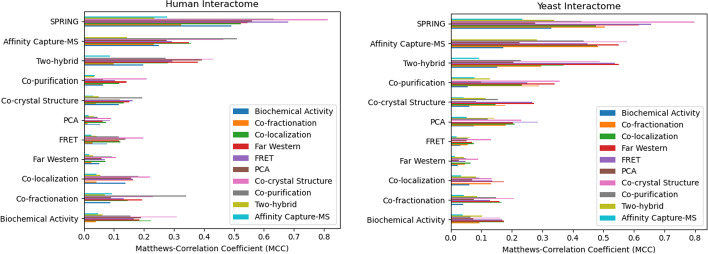
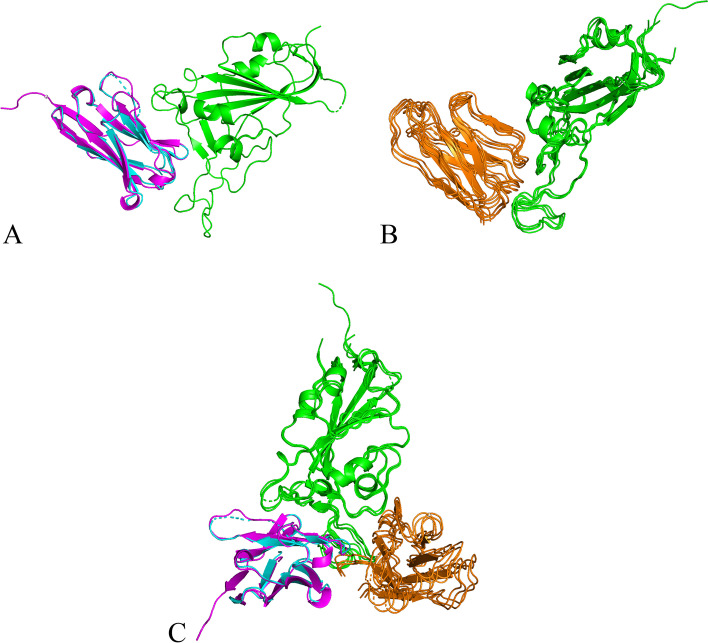
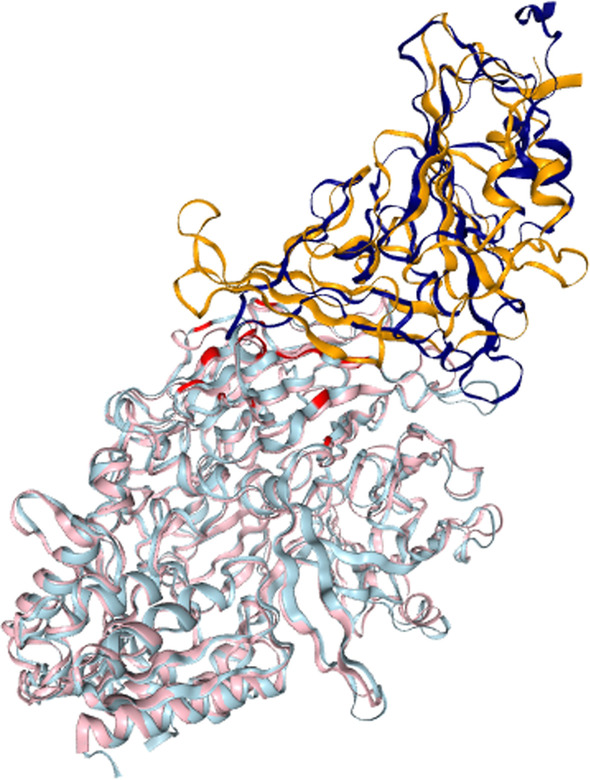
Application of our method to the Human and Yeast genomes yield protein-protein interaction networks similar in quality to common experimental methods. We identified and modeled Human proteins likely to interact with the papain-like protease of SARS-CoV2's non-structural protein 3. We also produced models of SARS-CoV2's spike protein (S) interacting with myelin-oligodendrocyte glycoprotein receptor and dipeptidyl peptidase-4.
The presented method is capable of confidently identifying interactions while providing high-quality multimeric structural models for experimental validation. The interactome modeling pipeline is available at usegalaxy.org and usegalaxy.eu.
蛋白质-蛋白质相互作用在几乎所有细胞过程中都起着至关重要的作用。鉴定相互作用的蛋白质可以深入了解生物,并为疾病治疗提供新的药物靶点。在这里,我们提出了一个公开的、自动化的流水线,用于预测全基因组蛋白质-蛋白质相互作用,并生成高质量的多聚体结构模型。
将我们的方法应用于人类和酵母基因组,得到的蛋白质-蛋白质相互作用网络的质量与常见的实验方法相似。我们鉴定并建模了可能与 SARS-CoV2 的非结构蛋白 3 的木瓜蛋白酶样蛋白酶相互作用的人类蛋白质。我们还生成了 SARS-CoV2 的刺突蛋白(S)与髓鞘少突胶质细胞糖蛋白受体和二肽基肽酶-4 相互作用的模型。
所提出的方法能够自信地识别相互作用,同时为实验验证提供高质量的多聚体结构模型。互作组建模流水线可在 usegalaxy.org 和 usegalaxy.eu 上使用。