Department of Plant Pathology, University of California, Davis, CA 95616.
Debre Zeit Agricultural Research Center, Ethiopian Institute for Agricultural Research, 32, Debre Zeit, Ethiopia.
Proc Natl Acad Sci U S A. 2023 Jul 4;120(27):e2220570120. doi: 10.1073/pnas.2220570120. Epub 2023 Jun 26.
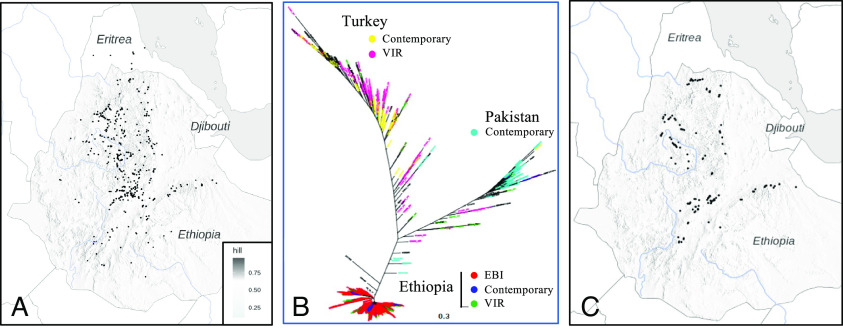
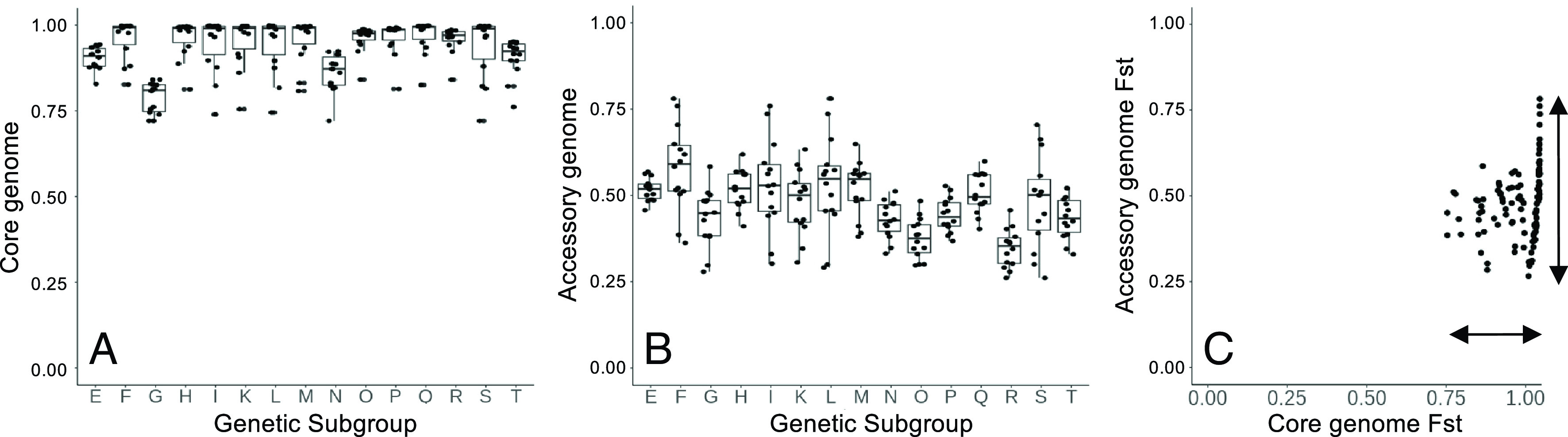
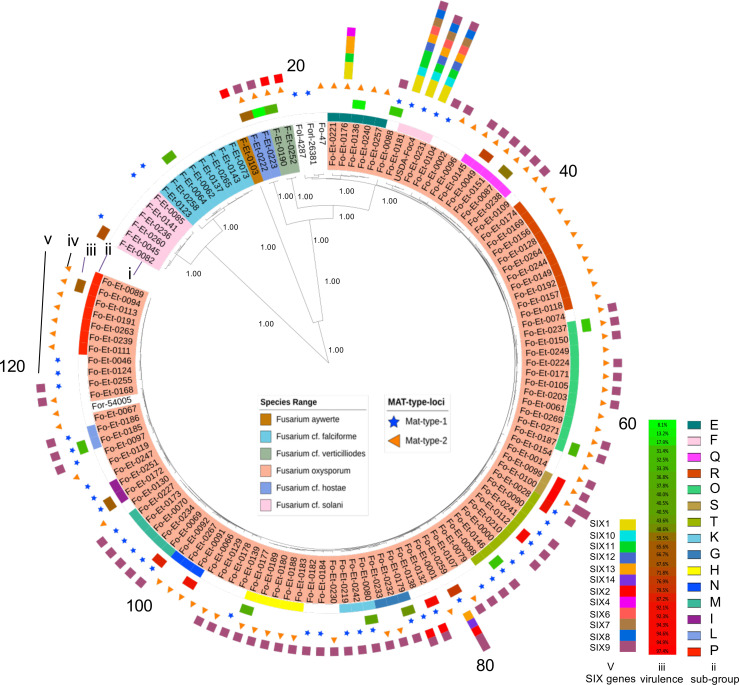
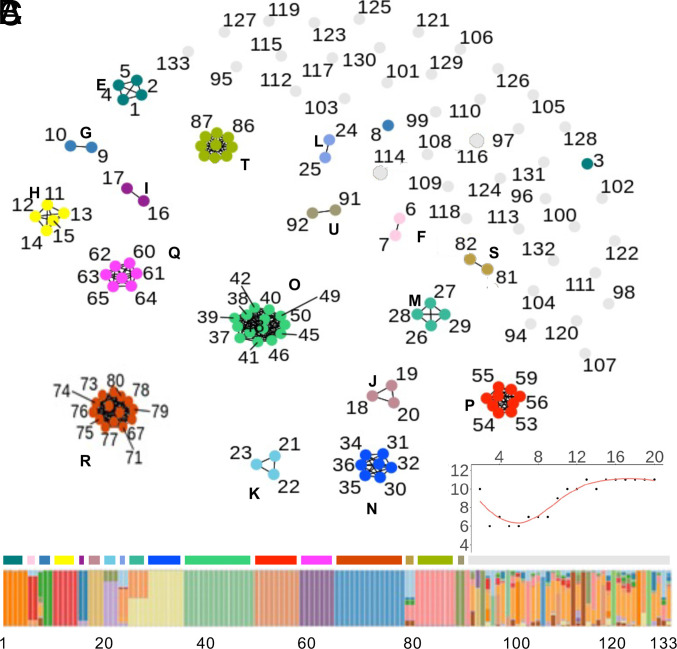
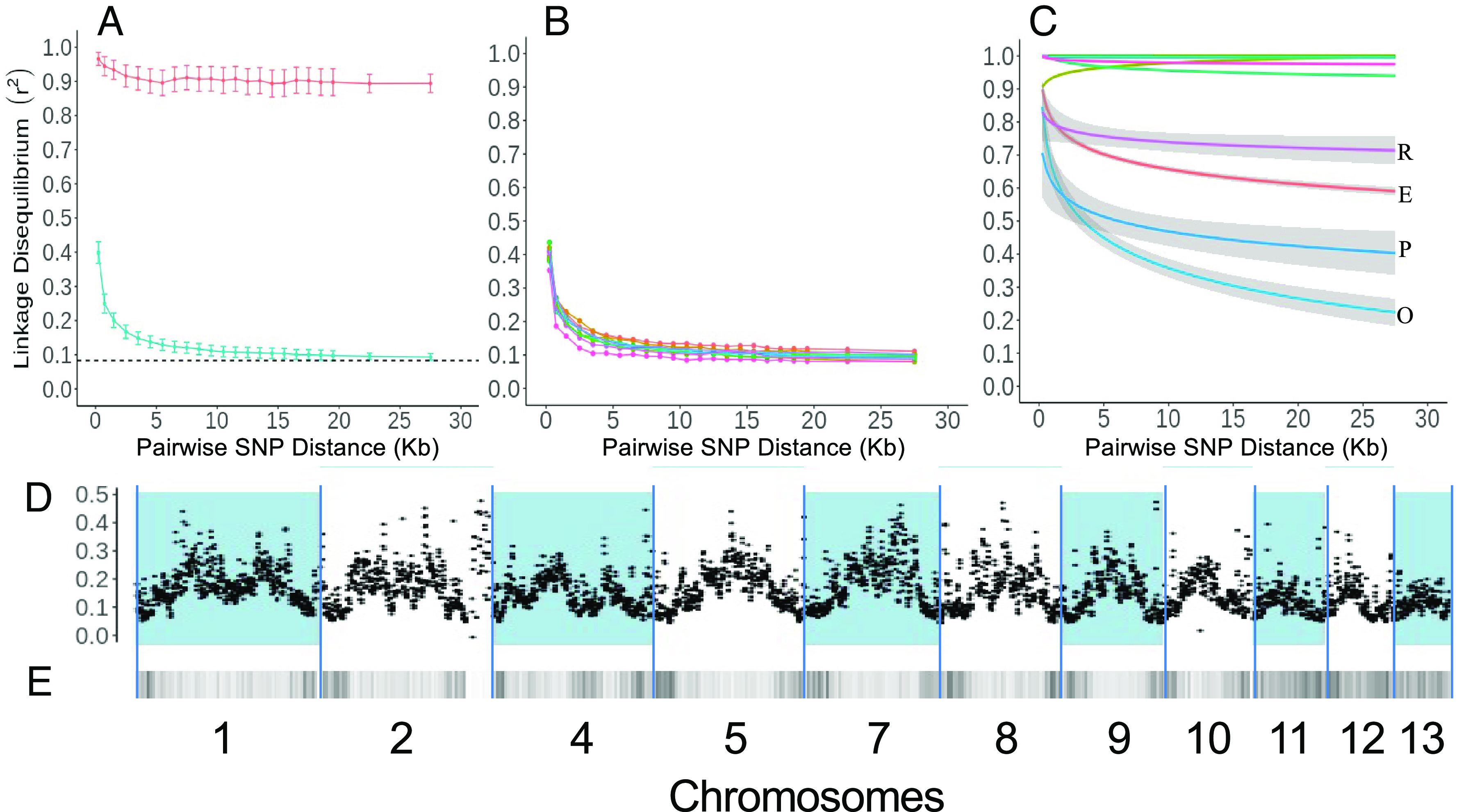
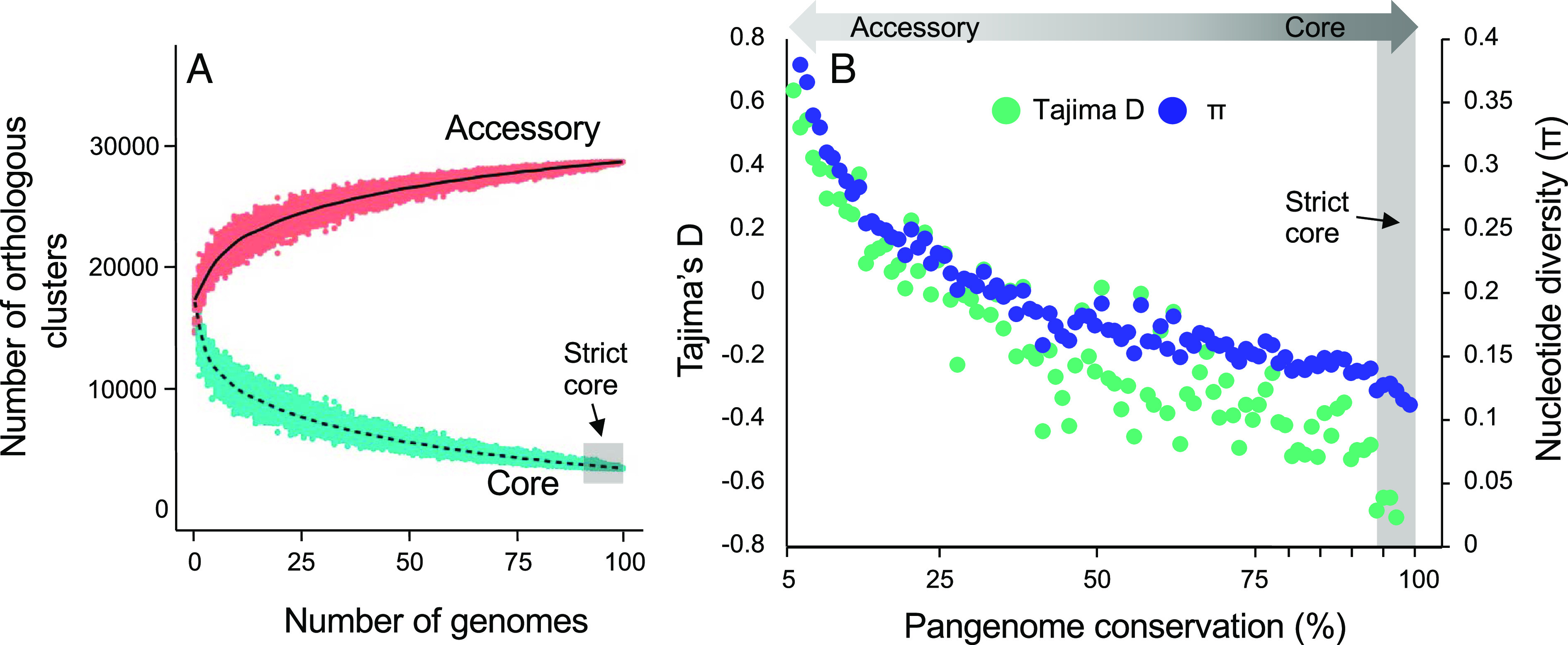
Understanding the origins of variation in agricultural pathogens is of fundamental interest and practical importance, especially for diseases that threaten food security. is among the most important of soil-borne pathogens, with a global distribution and an extensive host range. The pathogen is considered to be asexual, with horizontal transfer of chromosomes providing an analog of assortment by meiotic recombination. Here, we challenge those assumptions based on the results of population genomic analyses, describing the pathogen's diversity and inferring its origins and functional consequences in the context of a single, long-standing agricultural system. We identify simultaneously low nucleotide distance among strains, and unexpectedly high levels of genetic and genomic variability. We determine that these features arise from a combination of genome-scale recombination, best explained by widespread sexual reproduction, and presence-absence variation consistent with chromosomal rearrangement. Pangenome analyses document an accessory genome more than twice the size of the core genome, with contrasting evolutionary dynamics. The core genome is stable, with low diversity and high genetic differentiation across geographic space, while the accessory genome is paradoxically more diverse and unstable but with lower genetic differentiation and hallmarks of contemporary gene flow at local scales. We suggest a model in which episodic sexual reproduction generates haplotypes that are selected and then maintained through clone-like dynamics, followed by contemporary genomic rearrangements that reassort the accessory genome among sympatric strains. Taken together, these processes contribute unique genome content, including reassortment of virulence determinants that may explain observed variation in pathogenic potential.
了解农业病原体变异的起源具有根本的兴趣和重要的实际意义,特别是对于那些威胁粮食安全的疾病。茄青枯雷尔氏菌是最重要的土传病原体之一,具有全球分布和广泛的宿主范围。该病原体被认为是无性的,染色体的水平转移提供了减数分裂重组的类似选择。在这里,我们根据群体基因组分析的结果挑战了这些假设,描述了病原体的多样性,并在单个长期存在的农业系统背景下推断了其起源和功能后果。我们同时确定了菌株之间核苷酸距离低,而遗传和基因组变异水平高,这令人惊讶。我们确定这些特征是由基因组规模的重组以及广泛的有性生殖引起的,而存在缺失变异则与染色体重排一致。泛基因组分析记录了一个附加基因组,其大小是核心基因组的两倍多,具有相反的进化动态。核心基因组是稳定的,在地理空间中具有低多样性和高遗传分化,而附加基因组则相反,具有更高的多样性和不稳定性,但遗传分化较低,并且具有当代基因流的特征。我们提出了一个模型,即间歇性有性生殖产生单倍型,然后通过类似克隆的动态选择和维持,然后是当代基因组重排,在同域菌株之间重新分配附加基因组。这些过程共同产生了独特的基因组内容,包括毒力决定因素的重组,这可能解释了观察到的致病性潜力的变异。