Jiangxi Provincial Key Laboratory for Animal Health, Institute of Animal Population Health, College of Animal Science and Technology, Jiangxi Agricultural University, Nanchang, 330045, People's Republic of China.
Guangxi Vocational University of Agriculture, Nanning, 530007, Guangxi, China.
BMC Microbiol. 2023 Jul 7;23(1):180. doi: 10.1186/s12866-023-02925-7.
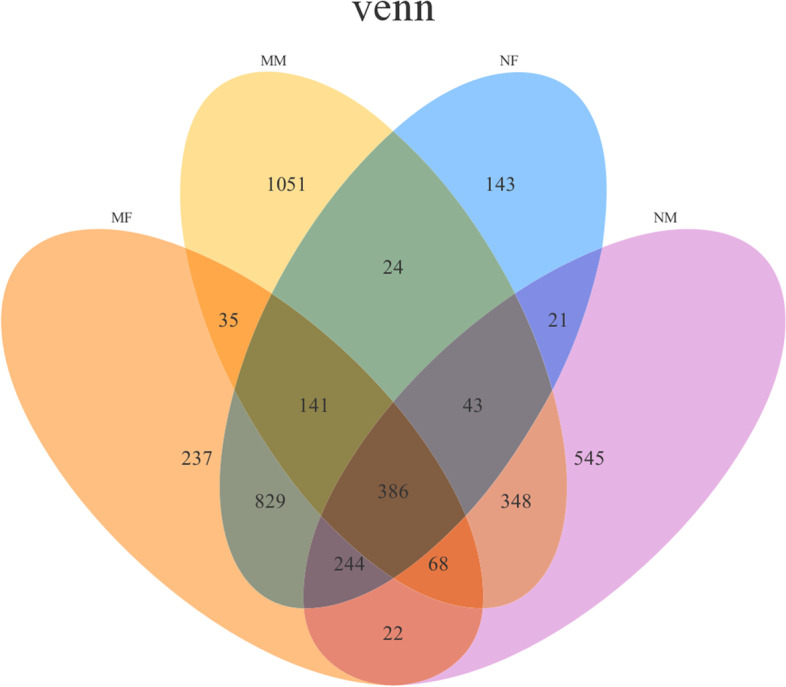
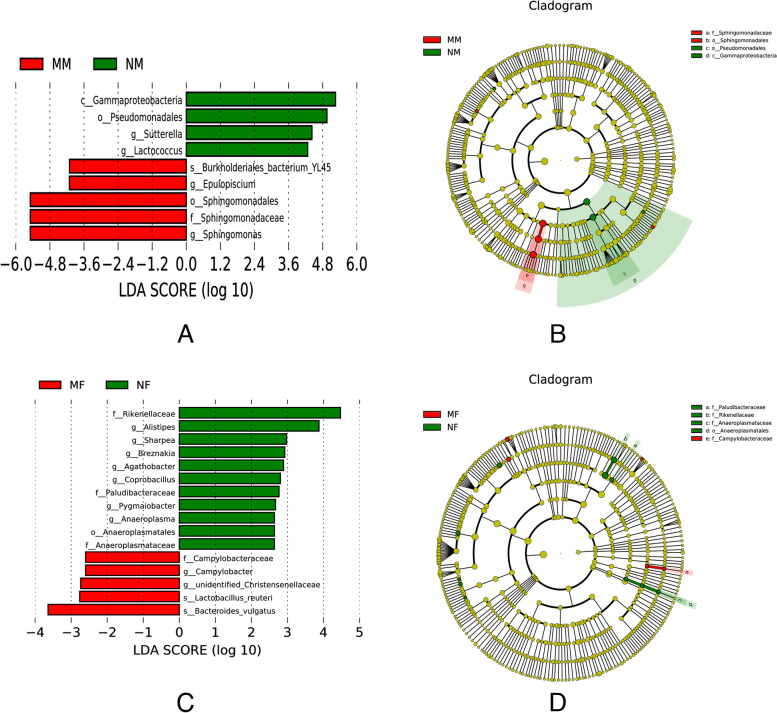
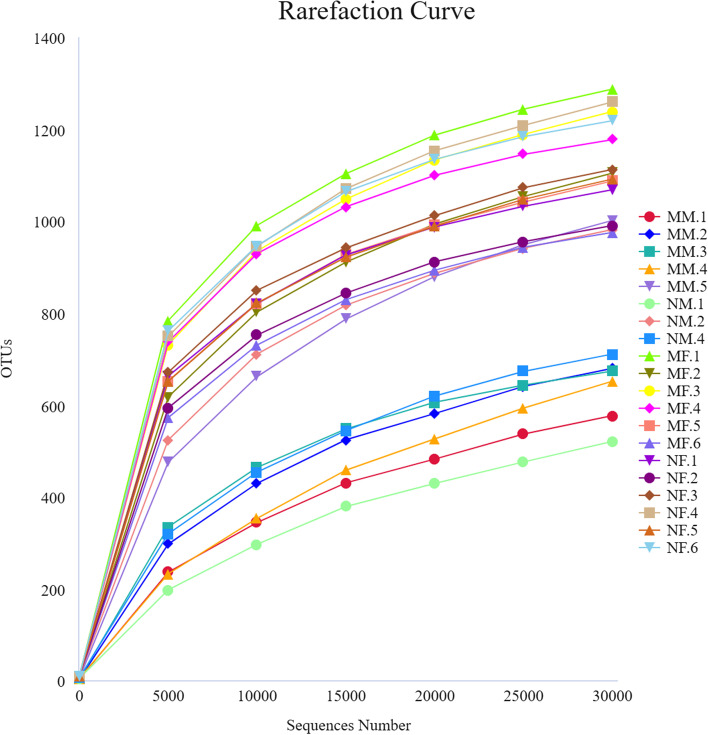
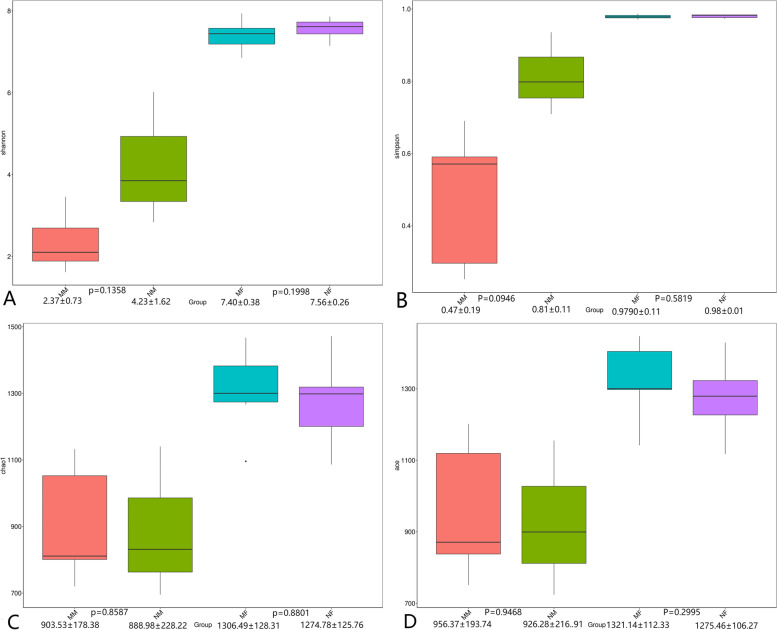
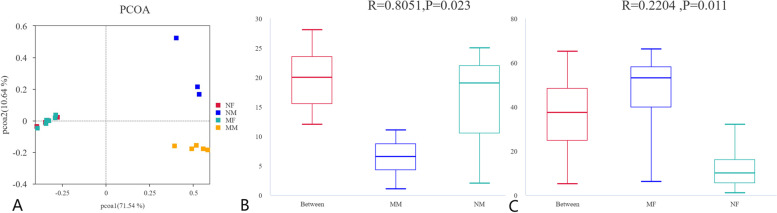
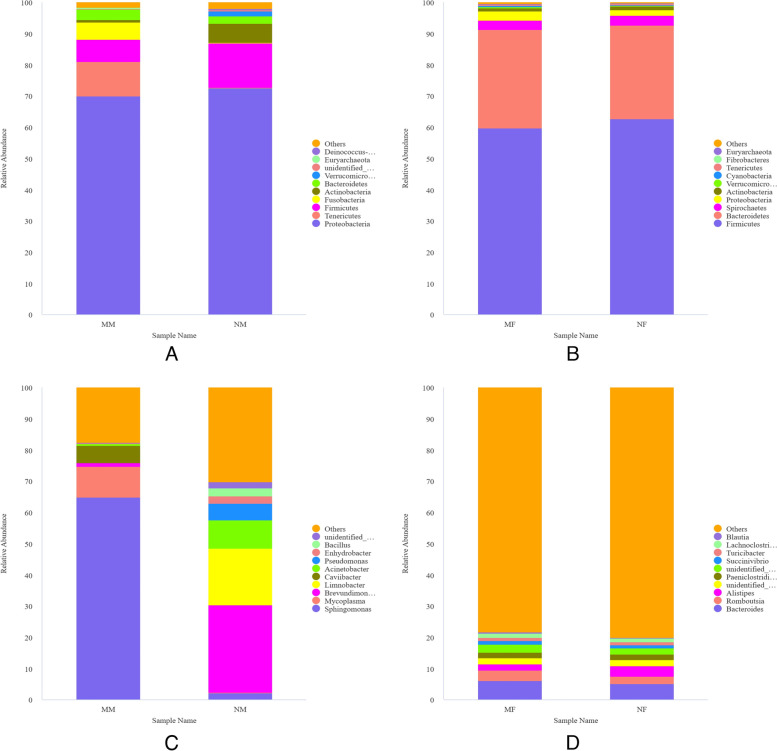
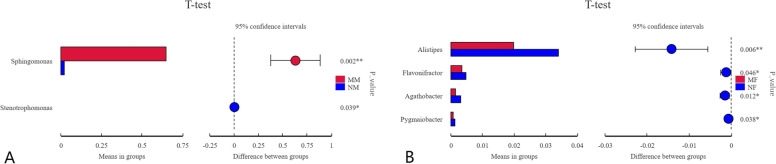
This study aimed to understand the changes in the milk and gut microbiota of dairy cows with mastitis, and to further explore the relationship between mastitis and the microbiota. In this study, we extracted microbial DNA from healthy and mastitis cows and performed high-throughput sequencing using the Illumina NovaSeq sequencing platform. OTU clustering was performed to analyze complexity, multi-sample comparisons, differences in community structure between groups, and differential analysis of species composition and abundance. The results showed that there were differences in microbial diversity and community composition in the milk and feces of normal and mastitis cows, where the diversity of microbiota decreased and species abundance increased in the mastitis group. There was a significant difference in the flora composition of the two groups of samples (P < 0.05), especially at the genus level, the difference in the milk samples was Sphingomonas (P < 0.05) and Stenotrophomonas (P < 0.05), the differences in stool samples were Alistipes (P < 0.05), Flavonifractor (P < 0.05), Agathobacter (P < 0.05) and Pygmaiobacter (P < 0.05). In conclusion, the microbiota of the udder and intestinal tissues of dairy cows suffering from mastitis will change significantly. This suggests that the development of mastitis is related to the endogenous pathway of microbial intestinal mammary glands, but the mechanisms involved need further study.
本研究旨在了解乳腺炎奶牛的奶和肠道微生物群的变化,并进一步探索乳腺炎与微生物群之间的关系。在这项研究中,我们从健康和乳腺炎奶牛中提取微生物 DNA,并使用 Illumina NovaSeq 测序平台进行高通量测序。进行了 OTU 聚类分析以分析复杂性、多样本比较、组间群落结构差异以及物种组成和丰度的差异分析。结果表明,正常和乳腺炎奶牛的奶和粪便中的微生物多样性和群落组成存在差异,其中乳腺炎组的微生物多样性降低,物种丰度增加。两组样本的菌群组成存在显著差异(P<0.05),特别是在属水平上,奶样中的差异是 Sphingomonas(P<0.05)和 Stenotrophomonas(P<0.05),粪便样本中的差异是 Alistipes(P<0.05)、Flavonifractor(P<0.05)、Agathobacter(P<0.05)和 Pygmaiobacter(P<0.05)。总之,患有乳腺炎的奶牛的乳房和肠道组织中的微生物群会发生明显变化。这表明乳腺炎的发展与微生物肠道乳腺的内源性途径有关,但需要进一步研究涉及的机制。