Linderstrøm-Lang Centre for Protein Science, Department of Biology, University of Copenhagen, Copenhagen, Denmark.
Nat Commun. 2023 Jul 13;14(1):4175. doi: 10.1038/s41467-023-39909-0.
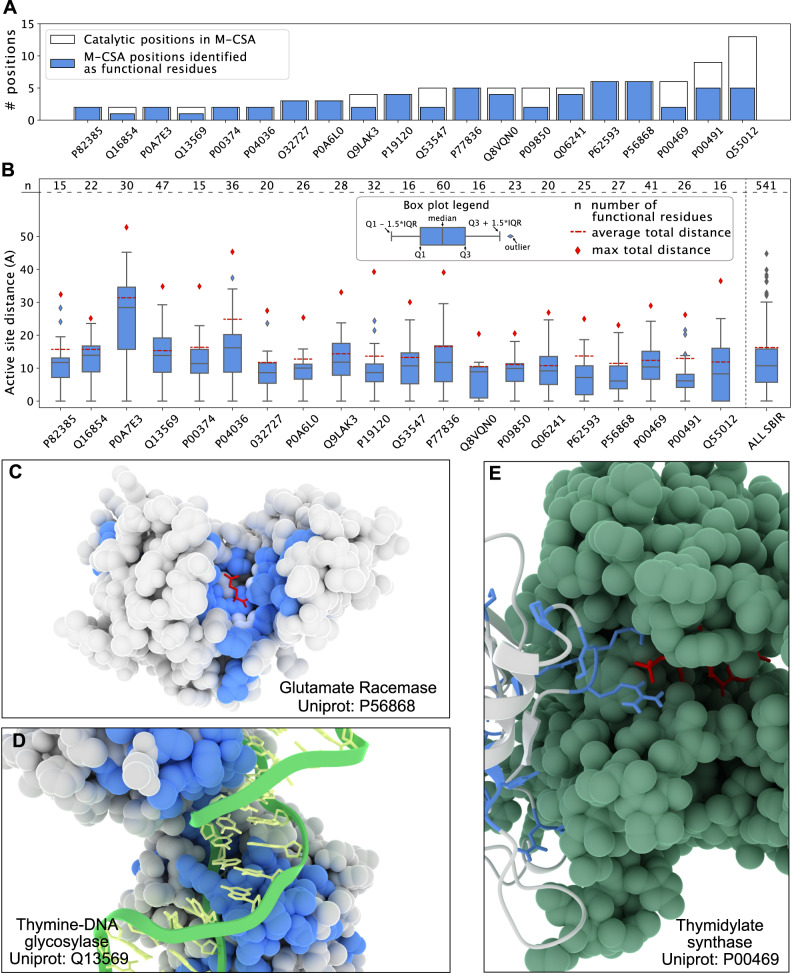
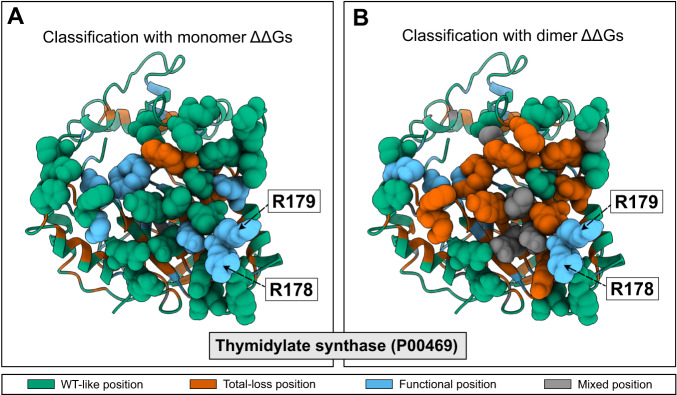
Proteins play important roles in biology, biotechnology and pharmacology, and missense variants are a common cause of disease. Discovering functionally important sites in proteins is a central but difficult problem because of the lack of large, systematic data sets. Sequence conservation can highlight residues that are functionally important but is often convoluted with a signal for preserving structural stability. We here present a machine learning method to predict functional sites by combining statistical models for protein sequences with biophysical models of stability. We train the model using multiplexed experimental data on variant effects and validate it broadly. We show how the model can be used to discover active sites, as well as regulatory and binding sites. We illustrate the utility of the model by prospective prediction and subsequent experimental validation on the functional consequences of missense variants in HPRT1 which may cause Lesch-Nyhan syndrome, and pinpoint the molecular mechanisms by which they cause disease.
蛋白质在生物学、生物技术和药理学中发挥着重要作用,错义变体是疾病的常见原因。由于缺乏大型系统数据集,发现蛋白质中的功能重要位点是一个核心但困难的问题。序列保守性可以突出功能重要的残基,但通常与保留结构稳定性的信号交织在一起。我们在这里提出了一种机器学习方法,通过将蛋白质序列的统计模型与稳定性的生物物理模型相结合,来预测功能位点。我们使用关于变体效应的多路实验数据来训练模型,并广泛验证它。我们展示了如何使用该模型来发现活性位点,以及调节和结合位点。我们通过前瞻性预测和随后对 HPRT1 中可能导致 Lesch-Nyhan 综合征的错义变体的功能后果的实验验证说明了该模型的实用性,并指出了它们导致疾病的分子机制。