School of Pharmacy, University College London, 29-39 Brunswick Square, London WC1N 1AX, UK.
Diamond Light Source, Harwell Science and Innovation Campus, Chilton, Didcot OX11 0DE, UK.
Int J Mol Sci. 2023 Jul 7;24(13):11197. doi: 10.3390/ijms241311197.
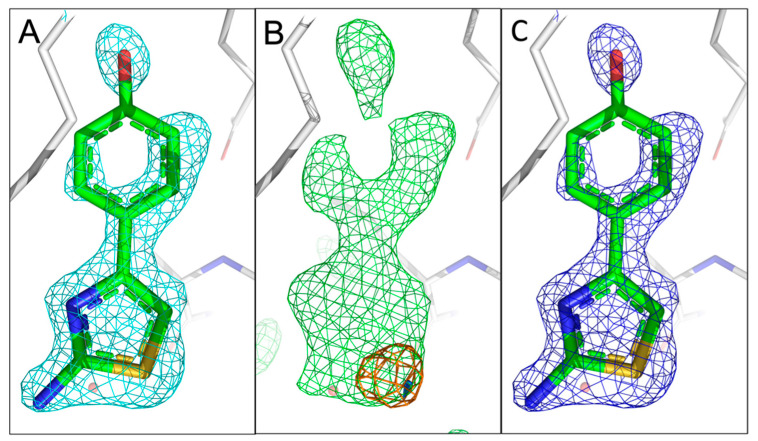
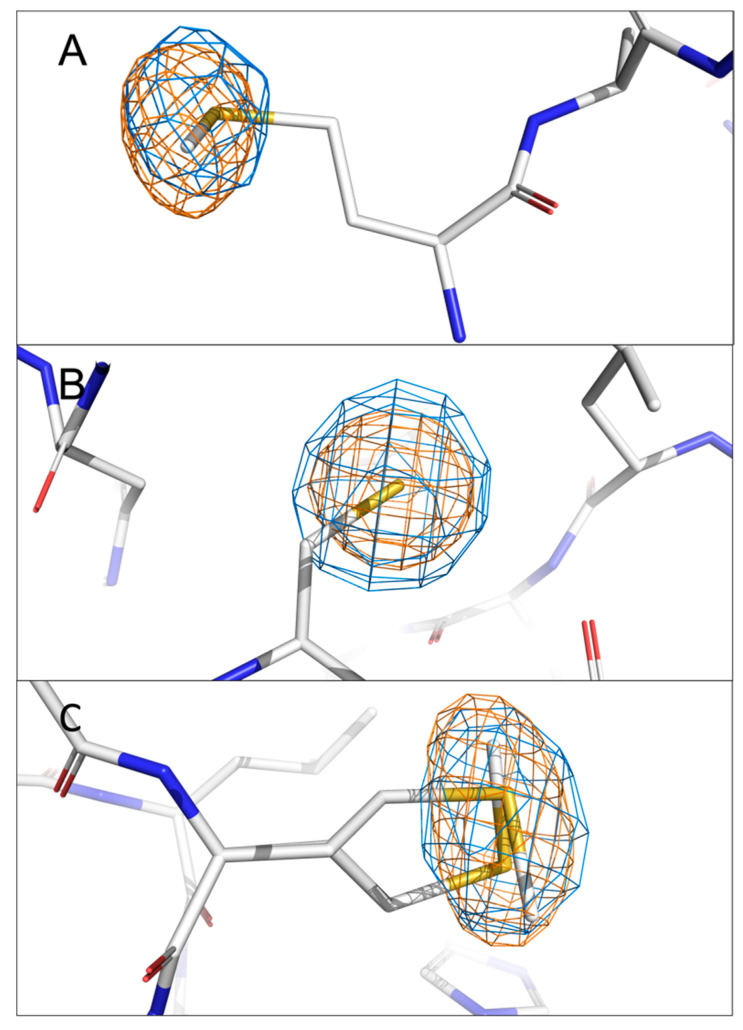
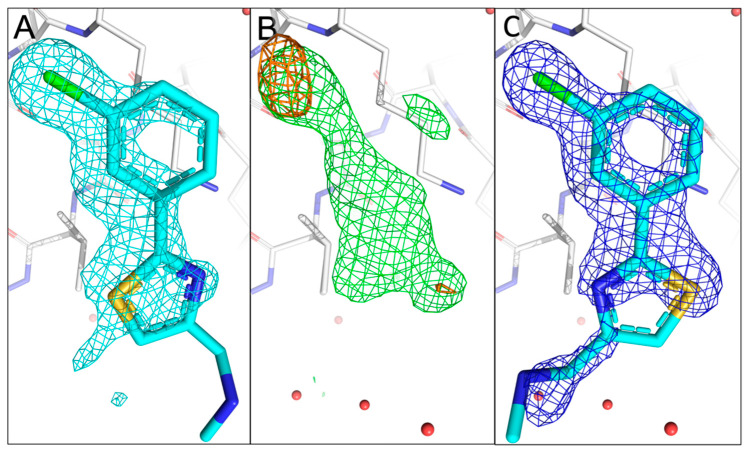
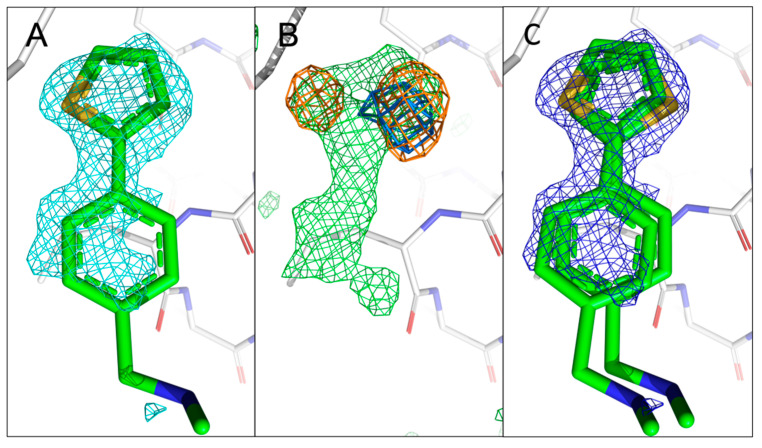
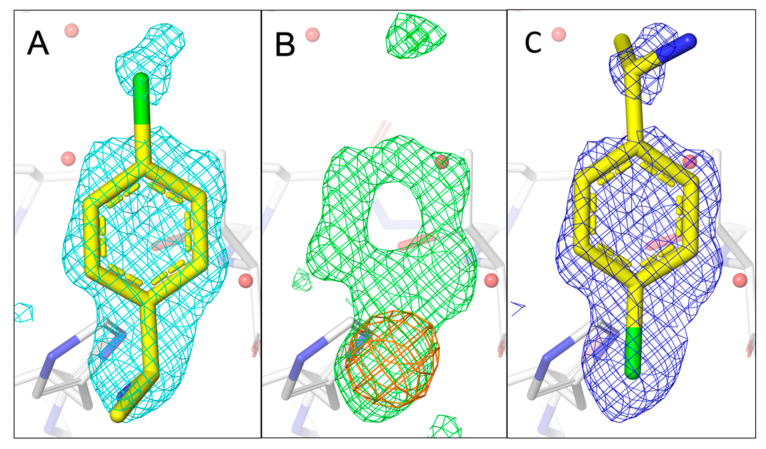
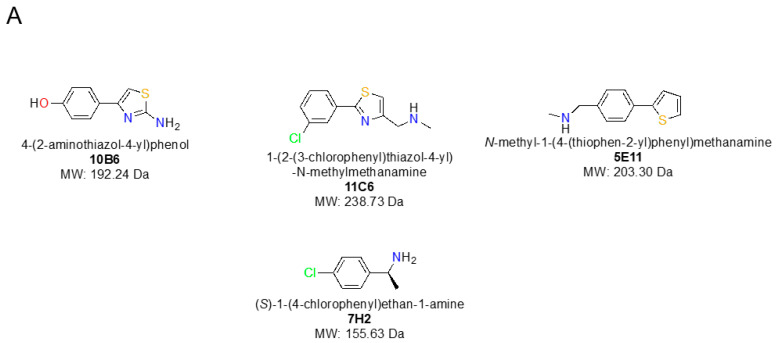
The identification of multiple simultaneous orientations of small molecule inhibitors binding to a protein target is a common challenge. It has recently been reported that the conformational heterogeneity of ligands is widely underreported in the Protein Data Bank, which is likely to impede optimal exploitation to improve affinity of these ligands. Significantly less is even known about multiple binding orientations for fragments (<300 Da), although this information would be essential for subsequent fragment optimisation using growing, linking or merging and rational structure-based design. Here, we use recently reported fragment hits for the SARS-CoV-2 non-structural protein 1 (nsp1) N-terminal domain to propose a general procedure for unambiguously identifying binding orientations of 2-dimensional fragments containing either sulphur or chloro substituents within the wavelength range of most tunable beamlines. By measuring datasets at two energies, using a tunable beamline operating in vacuum and optimised for data collection at very low X-ray energies, we show that the anomalous signal can be used to identify multiple orientations in small fragments containing sulphur and/or chloro substituents or to verify recently reported conformations. Although in this specific case we identified the positions of sulphur and chlorine in fragments bound to their protein target, we are confident that this work can be further expanded to additional atoms or ions which often occur in fragments. Finally, our improvements in the understanding of binding orientations will also serve to improve the rational optimisation of SARS-CoV-2 nsp1 fragment hits.
鉴定小分子抑制剂与蛋白质靶标结合的多个同时取向是一个常见的挑战。最近有报道称,配体的构象异质性在蛋白质数据库中被广泛低估,这可能会阻碍对这些配体的最佳利用以提高亲和力。对于片段(<300Da)的多个结合取向,人们甚至知之甚少,尽管对于使用生长、连接或合并和合理的基于结构的设计来进行后续片段优化,这种信息是必不可少的。在这里,我们使用最近报道的 SARS-CoV-2 非结构蛋白 1(nsp1)N 端结构域的片段命中来提出一种通用程序,用于明确鉴定在大多数可调谐光束线的波长范围内含有硫或氯取代基的二维片段的结合取向。通过在两个能量下测量数据集,使用在真空中运行的可调谐光束线并针对非常低的 X 射线能量进行数据收集进行优化,我们表明可以使用异常信号来识别含有硫和/或氯取代基的小片段中的多个取向,或验证最近报道的构象。虽然在这种特定情况下,我们确定了与蛋白质靶标结合的片段中硫和氯的位置,但我们有信心可以将这项工作扩展到其他经常出现在片段中的原子或离子。最后,我们对结合取向的理解的提高也将有助于提高对 SARS-CoV-2 nsp1 片段命中的合理优化。