Division of Biology and Biological Engineering, California Institute of Technology, Pasadena, California, United States of America.
Department of Computing and Mathematical Sciences, California Institute of Technology, Pasadena, California, United States of America.
PLoS Comput Biol. 2023 Aug 17;19(8):e1011288. doi: 10.1371/journal.pcbi.1011288. eCollection 2023 Aug.
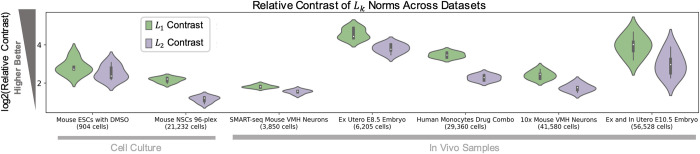
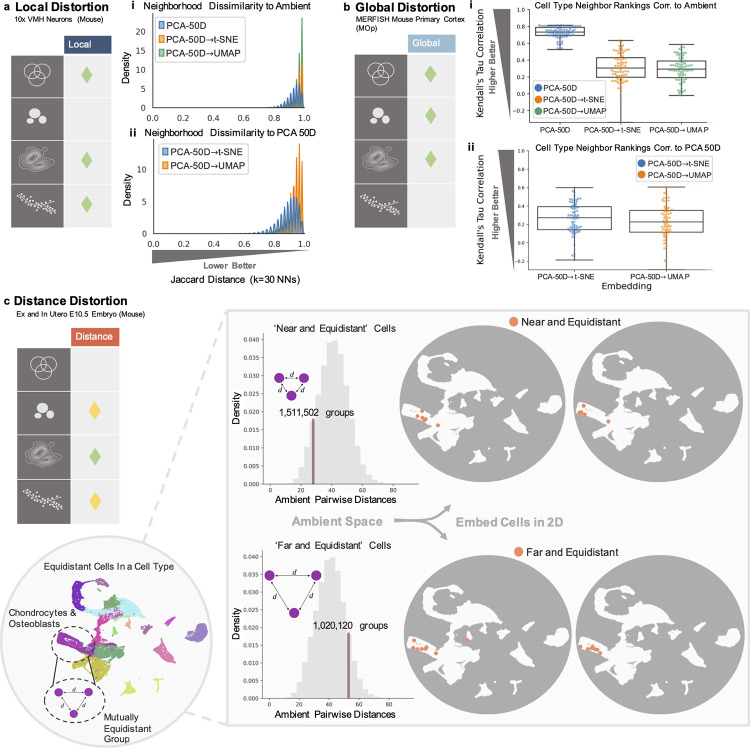
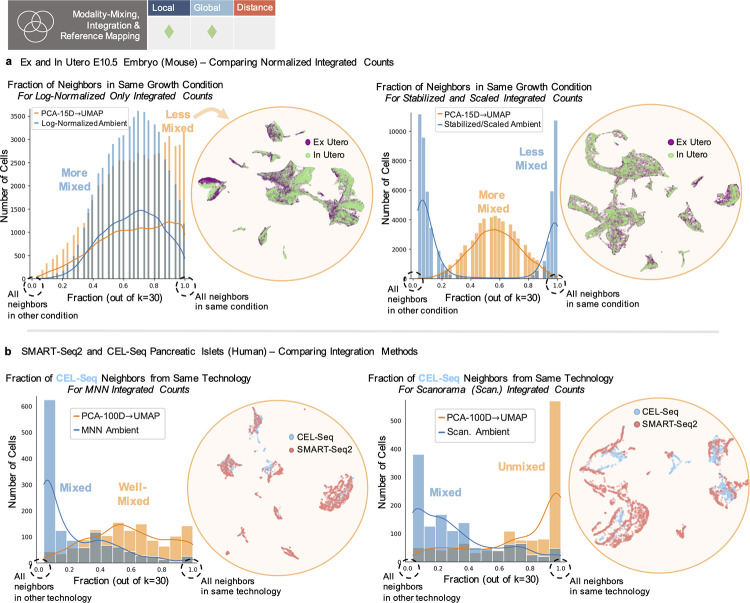
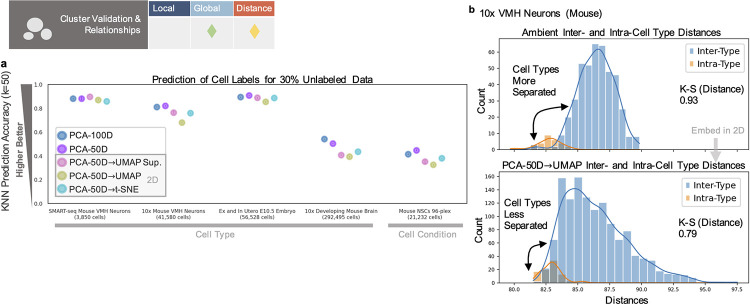
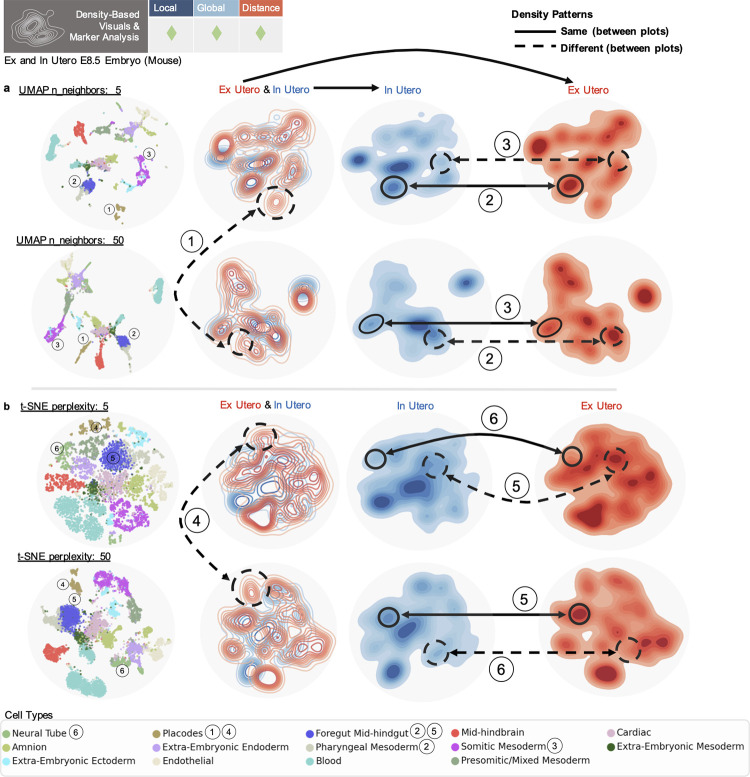
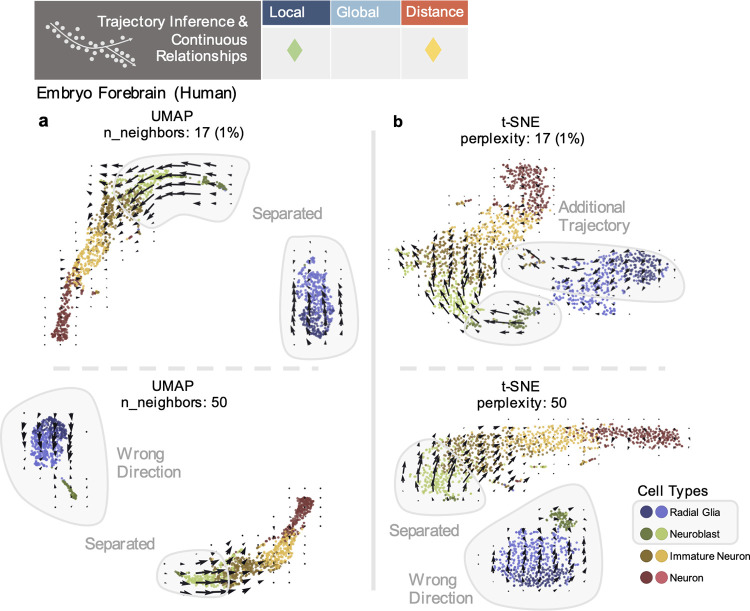
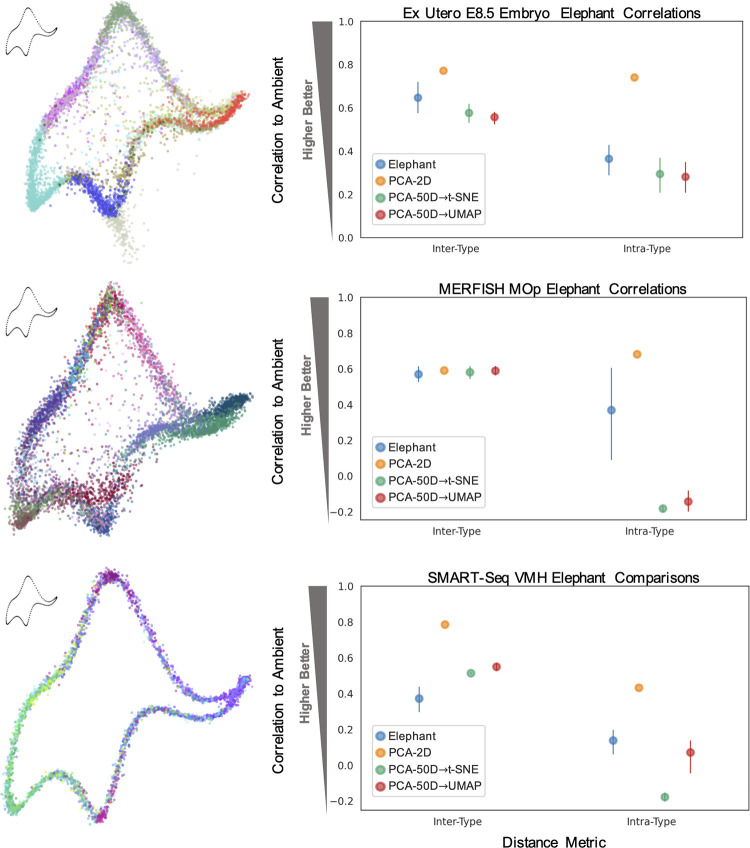
Dimensionality reduction is standard practice for filtering noise and identifying relevant features in large-scale data analyses. In biology, single-cell genomics studies typically begin with reduction to 2 or 3 dimensions to produce "all-in-one" visuals of the data that are amenable to the human eye, and these are subsequently used for qualitative and quantitative exploratory analysis. However, there is little theoretical support for this practice, and we show that extreme dimension reduction, from hundreds or thousands of dimensions to 2, inevitably induces significant distortion of high-dimensional datasets. We therefore examine the practical implications of low-dimensional embedding of single-cell data and find that extensive distortions and inconsistent practices make such embeddings counter-productive for exploratory, biological analyses. In lieu of this, we discuss alternative approaches for conducting targeted embedding and feature exploration to enable hypothesis-driven biological discovery.
降维是过滤噪声和识别大规模数据分析中相关特征的标准做法。在生物学中,单细胞基因组学研究通常首先进行到 2 或 3 维,以生成数据的“一站式”可视化,这些可视化适合人眼观察,随后可用于定性和定量探索性分析。然而,这种做法几乎没有理论支持,我们表明,从数百或数千个维度到 2 个维度的极端降维不可避免地会对高维数据集造成严重的扭曲。因此,我们研究了单细胞数据低维嵌入的实际影响,发现广泛的扭曲和不一致的做法使得这种嵌入对探索性、生物学分析适得其反。为此,我们讨论了用于进行有针对性的嵌入和特征探索的替代方法,以支持基于假设的生物发现。