Channing Division of Network Medicine, Department of Medicine, Brigham and Women's Hospital and Harvard Medical School, Boston, MA, USA.
Qingdao OE Biotechnology Company Limited, Qingdao, Shandong, China.
Nat Commun. 2023 Sep 1;14(1):5321. doi: 10.1038/s41467-023-41099-8.
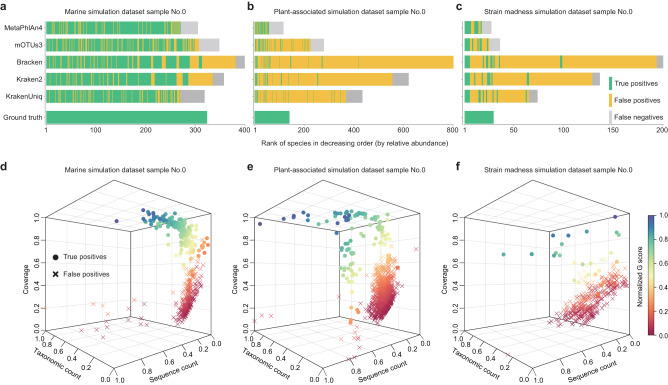
Accurate species identification and abundance estimation are critical for the interpretation of whole metagenome sequencing (WMS) data. Yet, existing metagenomic profilers suffer from false-positive identifications, which can account for more than 90% of total identified species. Here, by leveraging species-specific Type IIB restriction endonuclease digestion sites as reference instead of universal markers or whole microbial genomes, we present a metagenomic profiler, MAP2B (MetAgenomic Profiler based on type IIB restriction sites), to resolve those issues. We first illustrate the pitfalls of using relative abundance as the only feature in determining false positives. We then propose a feature set to distinguish false positives from true positives, and using simulated metagenomes from CAMI2, we establish a false-positive recognition model. By benchmarking the performance in metagenomic profiling using a simulation dataset with varying sequencing depth and species richness, we illustrate the superior performance of MAP2B over existing metagenomic profilers in species identification. We further test the performance of MAP2B using real WMS data from an ATCC mock community, confirming its superior precision against sequencing depth. Finally, by leveraging WMS data from an IBD cohort, we demonstrate the taxonomic features generated by MAP2B can better discriminate IBD and predict metabolomic profiles.
准确的物种鉴定和丰度估计对于解释全宏基因组测序(WMS)数据至关重要。然而,现有的宏基因组分析器存在假阳性鉴定问题,这些问题可能占总鉴定物种的 90%以上。在这里,我们利用物种特异性的 Type IIB 限制内切酶消化位点作为参考,而不是通用标记或整个微生物基因组,提出了一种宏基因组分析器 MAP2B(基于 Type IIB 限制位点的宏基因组分析器),以解决这些问题。我们首先说明了仅使用相对丰度作为确定假阳性的唯一特征所存在的问题。然后,我们提出了一个特征集来区分假阳性和真阳性,并使用来自 CAMI2 的模拟宏基因组建立了假阳性识别模型。通过使用具有不同测序深度和物种丰富度的模拟数据集来评估宏基因组分析的性能,我们说明了 MAP2B 在物种鉴定方面优于现有宏基因组分析器的性能。我们进一步使用来自 ATCC 模拟群落的真实 WMS 数据测试了 MAP2B 的性能,证实了其在对抗测序深度方面的卓越精度。最后,通过利用来自 IBD 队列的 WMS 数据,我们证明了 MAP2B 生成的分类特征可以更好地区分 IBD 并预测代谢组学特征。