Bioinformatics Interdepartmental Program, University of California, Los Angeles, Los Angeles, CA, 90095, USA.
Department of Biological Chemistry, University of California, Los Angeles, Los Angeles, CA, 90095, USA.
Genome Biol. 2023 Sep 7;24(1):203. doi: 10.1186/s13059-023-03041-5.
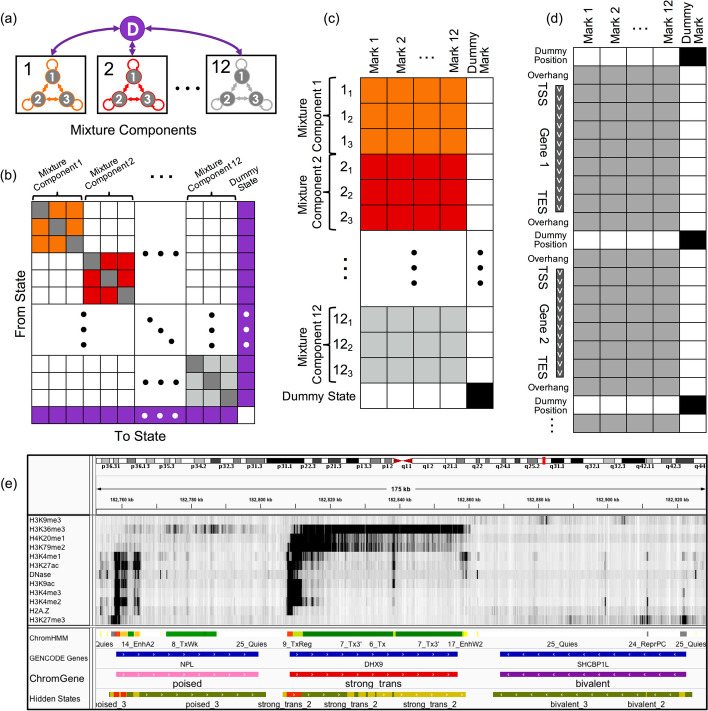
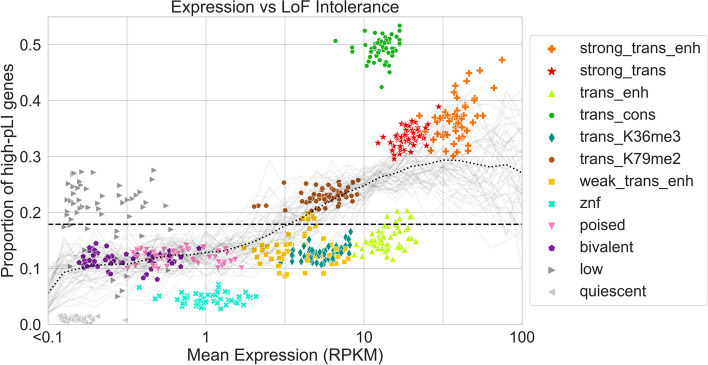
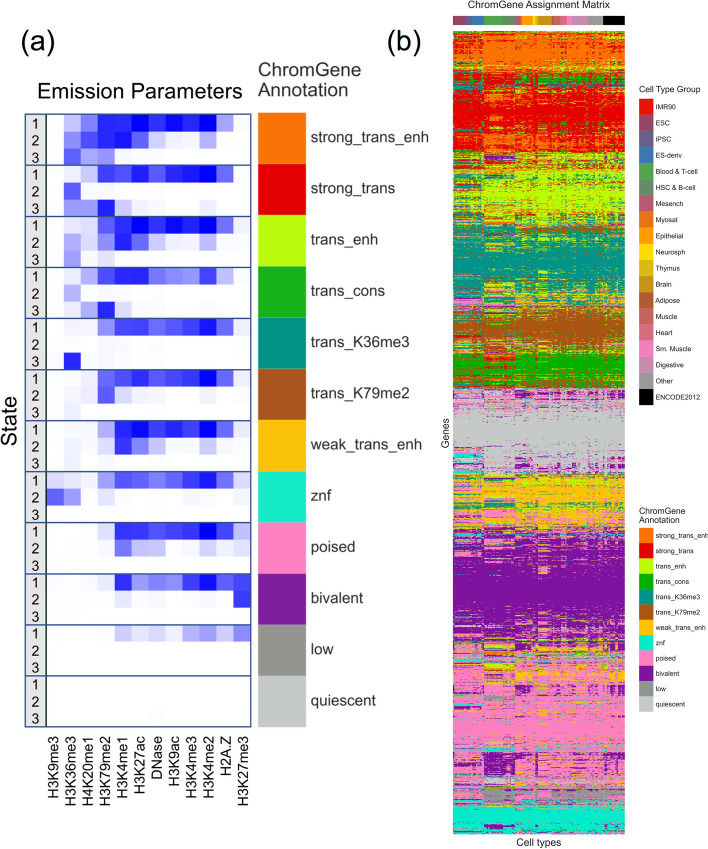
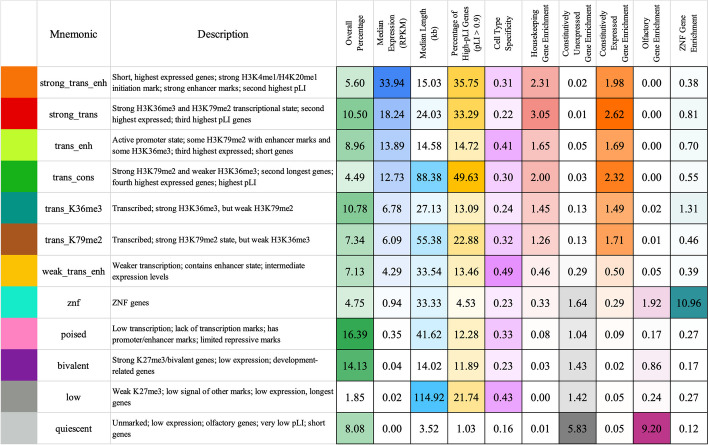
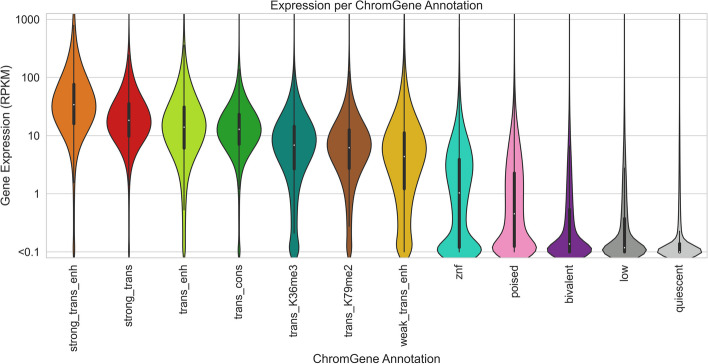
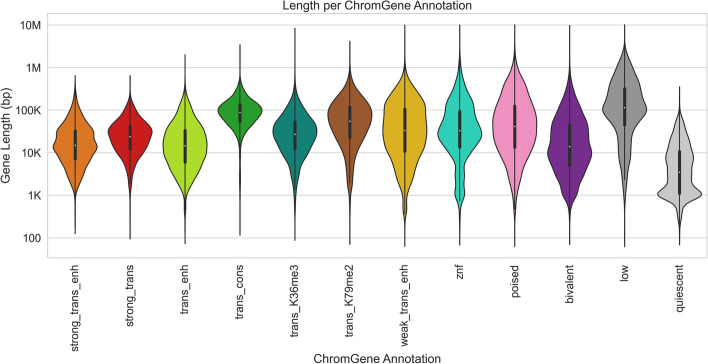
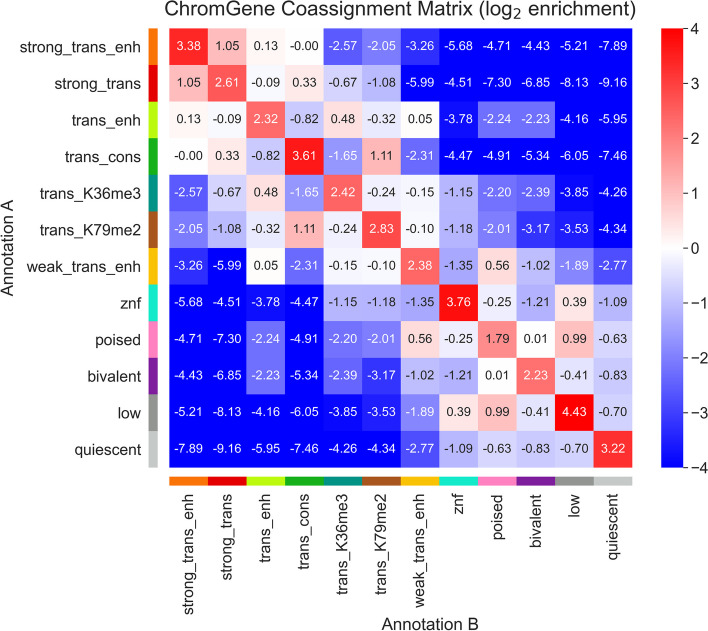
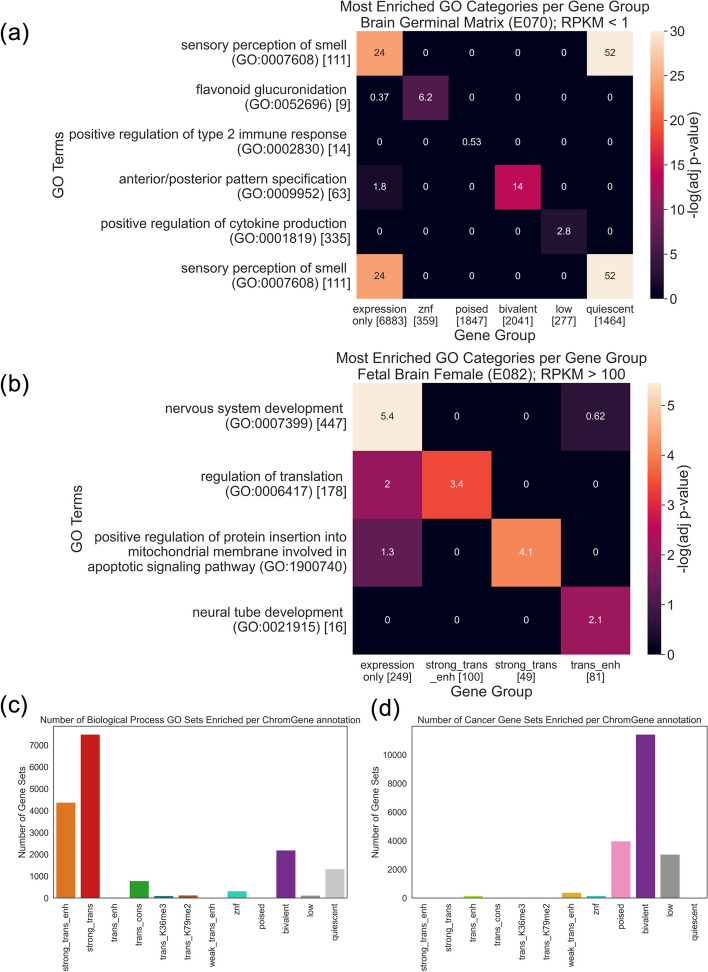
Various computational approaches have been developed to annotate epigenomes on a per-position basis by modeling combinatorial and spatial patterns within epigenomic data. However, such annotations are less suitable for gene-based analyses. We present ChromGene, a method based on a mixture of learned hidden Markov models, to annotate genes based on multiple epigenomic maps across the gene body and flanks. We provide ChromGene assignments for over 100 cell and tissue types. We characterize the mixture components in terms of gene expression, constraint, and other gene annotations. The ChromGene method and annotations will provide a useful resource for gene-based epigenomic analyses.
已经开发了各种计算方法来对每个位置的表观基因组进行注释,通过对表观基因组数据中的组合和空间模式进行建模。然而,这种注释不太适合基于基因的分析。我们提出了 ChromGene 方法,它基于学习的隐马尔可夫模型的混合模型,基于基因体和侧翼的多个表观基因组图谱来注释基因。我们提供了超过 100 种细胞和组织类型的 ChromGene 分配。我们根据基因表达、约束和其他基因注释来描述混合物成分。ChromGene 方法和注释将为基于基因的表观基因组分析提供有用的资源。