Botía Juan A, Vandrovcova Jana, Forabosco Paola, Guelfi Sebastian, D'Sa Karishma, Hardy John, Lewis Cathryn M, Ryten Mina, Weale Michael E
Department of Molecular Neuroscience, Institute of Neurology, University College London, Queen Square, London, WC1N, UK.
Department of Medical & Molecular Genetics, School of Medical Sciences, King's College London, Guy's Hospital, London, SE1 9RT, UK.
BMC Syst Biol. 2017 Apr 12;11(1):47. doi: 10.1186/s12918-017-0420-6.
Weighted Gene Co-expression Network Analysis (WGCNA) is a widely used R software package for the generation of gene co-expression networks (GCN). WGCNA generates both a GCN and a derived partitioning of clusters of genes (modules). We propose k-means clustering as an additional processing step to conventional WGCNA, which we have implemented in the R package km2gcn (k-means to gene co-expression network, https://github.com/juanbot/km2gcn ).
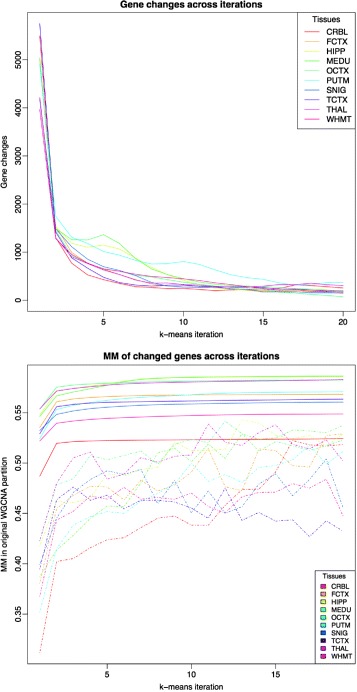
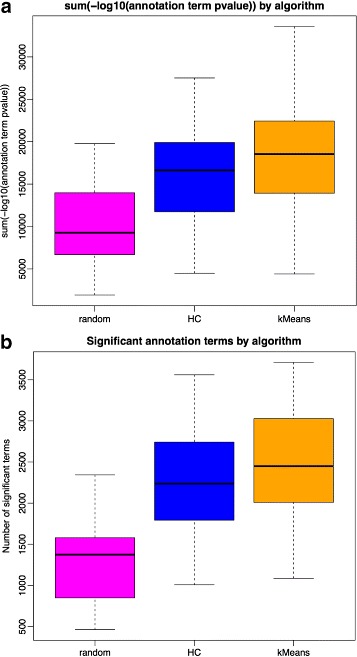
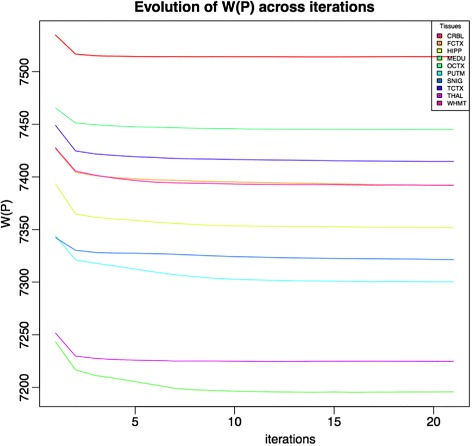
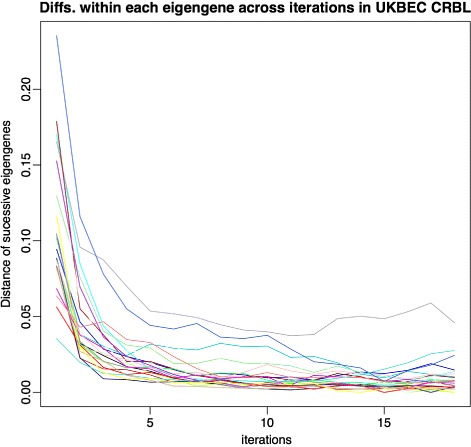
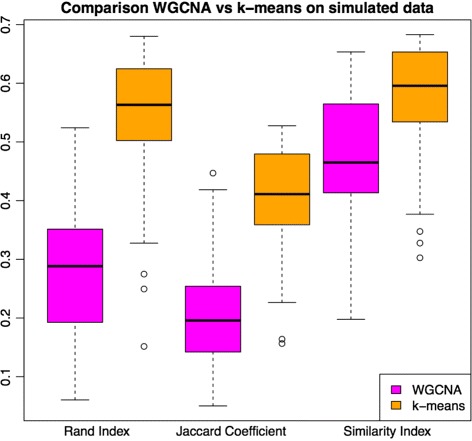
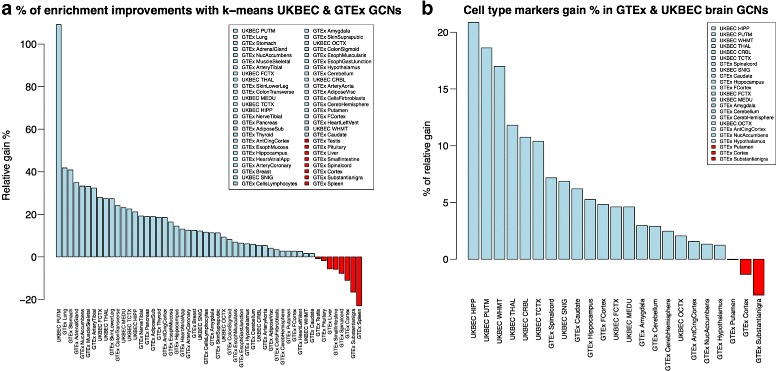
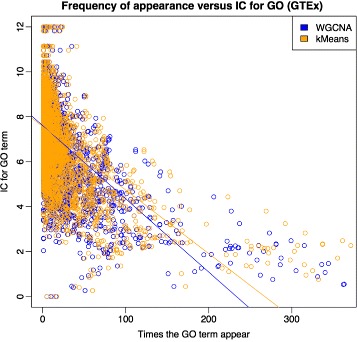
We assessed our method on networks created from UKBEC data (10 different human brain tissues), on networks created from GTEx data (42 human tissues, including 13 brain tissues), and on simulated networks derived from GTEx data. We observed substantially improved module properties, including: (1) few or zero misplaced genes; (2) increased counts of replicable clusters in alternate tissues (x3.1 on average); (3) improved enrichment of Gene Ontology terms (seen in 48/52 GCNs) (4) improved cell type enrichment signals (seen in 21/23 brain GCNs); and (5) more accurate partitions in simulated data according to a range of similarity indices.
The results obtained from our investigations indicate that our k-means method, applied as an adjunct to standard WGCNA, results in better network partitions. These improved partitions enable more fruitful downstream analyses, as gene modules are more biologically meaningful.
加权基因共表达网络分析(WGCNA)是一个广泛使用的R软件包,用于生成基因共表达网络(GCN)。WGCNA既生成一个GCN,也生成基因簇(模块)的派生划分。我们提出将k均值聚类作为传统WGCNA的一个额外处理步骤,我们已在R包km2gcn(从k均值到基因共表达网络,https://github.com/juanbot/km2gcn )中实现了这一方法。
我们在由UKBEC数据(10种不同的人类脑组织)创建的网络、由GTEx数据(42种人类组织,包括13种脑组织)创建的网络以及从GTEx数据派生的模拟网络上评估了我们的方法。我们观察到模块属性有显著改善,包括:(1)错误放置的基因很少或为零;(2)在其他组织中可复制簇的数量增加(平均增加3.1倍);(3)基因本体术语的富集得到改善(在52个GCN中的48个中可见);(4)细胞类型富集信号得到改善(在23个脑GCN中的21个中可见);以及(5)根据一系列相似性指标,模拟数据中的划分更准确。
我们的研究结果表明,我们的k均值方法作为标准WGCNA的辅助方法,可产生更好的网络划分。这些改进的划分使下游分析更有成效,因为基因模块更具生物学意义。