Data Analytics Research Laboratory (DARELab), Institute of Mathematical Sciences and Physics, University of the Philippines Los Baños, 4031, Los Baños, Laguna, Philippines.
Computational Interdisciplinary Research Laboratory (CINTERLabs), University of the Philippines Los Baños, 4031, Los Baños, Laguna, Philippines.
Sci Rep. 2023 Sep 14;13(1):15230. doi: 10.1038/s41598-023-41862-3.
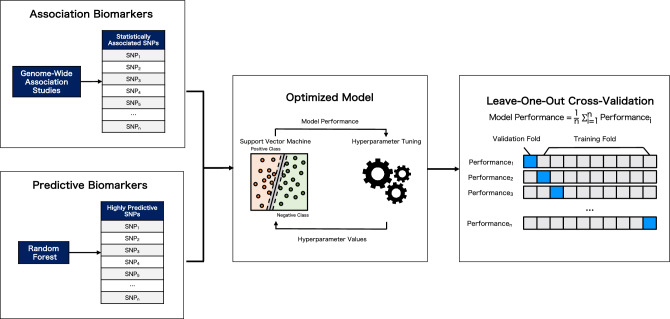
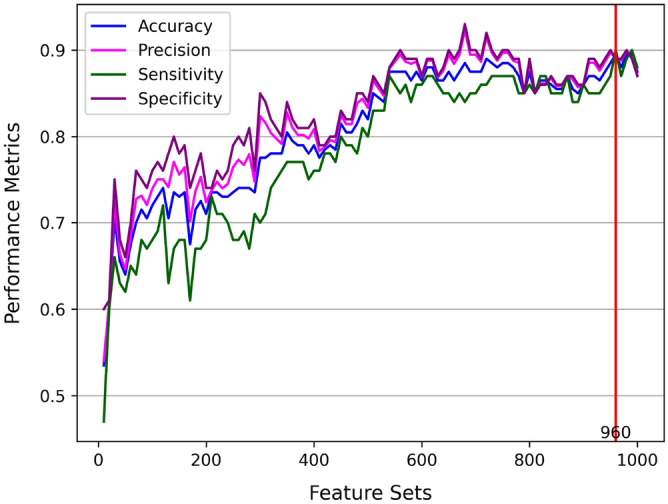
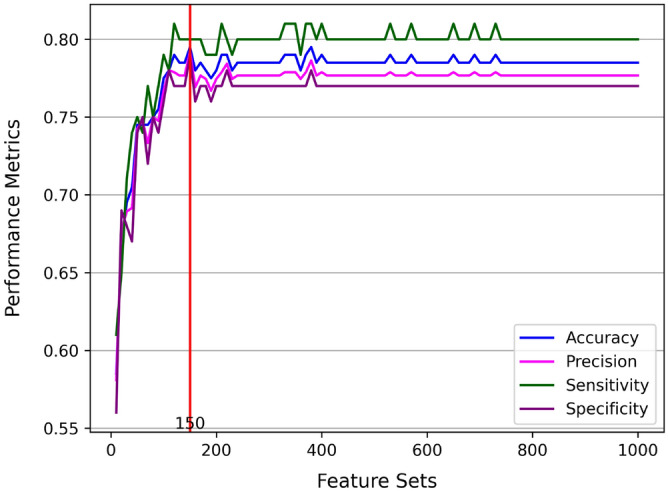
The genetic basis of phenotypic emergence provides valuable information for assessing individual risk. While association studies have been pivotal in identifying genetic risk factors within a population, complementing it with insights derived from predictions studies that assess individual-level risk offers a more comprehensive approach to understanding phenotypic expression. In this study, we established personalized risk assessment models using single-nucleotide polymorphism (SNP) data from 200 Korean patients, of which 100 experienced hepatitis B surface antigen (HBsAg) seroclearance and 100 patients demonstrated high levels of HBsAg. The risk assessment models determined the predictive power of the following: (1) genome-wide association study (GWAS)-identified candidate biomarkers considered significant in a reference study and (2) machine learning (ML)-identified candidate biomarkers with the highest feature importance scores obtained by using random forest (RF). While utilizing all features yielded 64% model accuracy, using relevant biomarkers achieved higher model accuracies: 82% for 52 GWAS-identified candidate biomarkers, 71% for three GWAS-identified biomarkers, and 80% for 150 ML-identified candidate biomarkers. Findings highlight that the joint contributions of relevant biomarkers significantly influence phenotypic emergence. On the other hand, combining ML-identified candidate biomarkers into the pool of GWAS-identified candidate biomarkers resulted in the improved predictive accuracy of 90%, demonstrating the capability of ML as an auxiliary analysis to GWAS. Furthermore, some of the ML-identified candidate biomarkers were found to be linked with hepatocellular carcinoma (HCC), reinforcing previous claims that HCC can still occur despite the absence of HBsAg.
表型出现的遗传基础为评估个体风险提供了有价值的信息。虽然关联研究在鉴定人群中的遗传风险因素方面发挥了重要作用,但通过评估个体风险的预测研究来补充这些研究提供了一种更全面的方法来理解表型表达。在这项研究中,我们使用来自 200 名韩国患者的单核苷酸多态性 (SNP) 数据建立了个性化风险评估模型,其中 100 名患者经历了乙型肝炎表面抗原 (HBsAg) 血清清除,100 名患者表现出 HBsAg 高水平。风险评估模型确定了以下两种情况的预测能力:(1)全基因组关联研究 (GWAS) 确定的候选生物标志物在参考研究中被认为具有显著意义,(2)机器学习 (ML) 确定的候选生物标志物,这些标志物通过使用随机森林 (RF) 获得了最高的特征重要性评分。虽然使用所有特征可以达到 64%的模型准确性,但使用相关生物标志物可以获得更高的模型准确性:52 个 GWAS 确定的候选生物标志物的准确性为 82%,3 个 GWAS 确定的生物标志物的准确性为 71%,150 个 ML 确定的候选生物标志物的准确性为 80%。研究结果表明,相关生物标志物的共同贡献显著影响表型出现。另一方面,将 ML 确定的候选生物标志物与 GWAS 确定的候选生物标志物结合在一起,可以将预测准确性提高到 90%,这表明 ML 作为 GWAS 的辅助分析具有一定的能力。此外,一些 ML 确定的候选生物标志物与肝细胞癌 (HCC) 有关,这进一步证实了尽管没有 HBsAg,HCC 仍然可能发生的说法。