Computational Science Laboratory, Universitat Pompeu Fabra, Barcelona Biomedical Research Park (PRBB), Carrer Dr. Aiguader 88, 08003, Barcelona, Spain.
Acellera Labs, Doctor Trueta 183, 08005, Barcelona, Spain.
Nat Commun. 2023 Sep 15;14(1):5739. doi: 10.1038/s41467-023-41343-1.
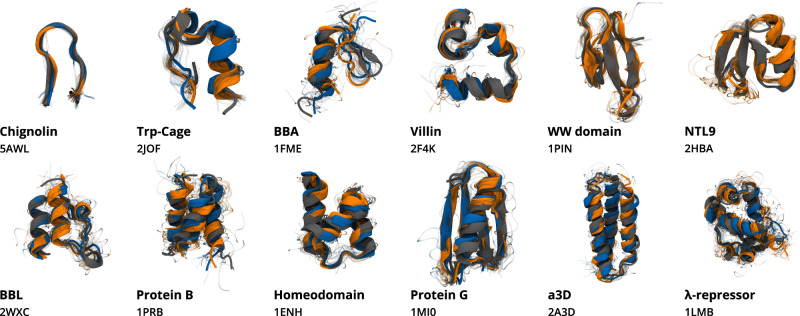
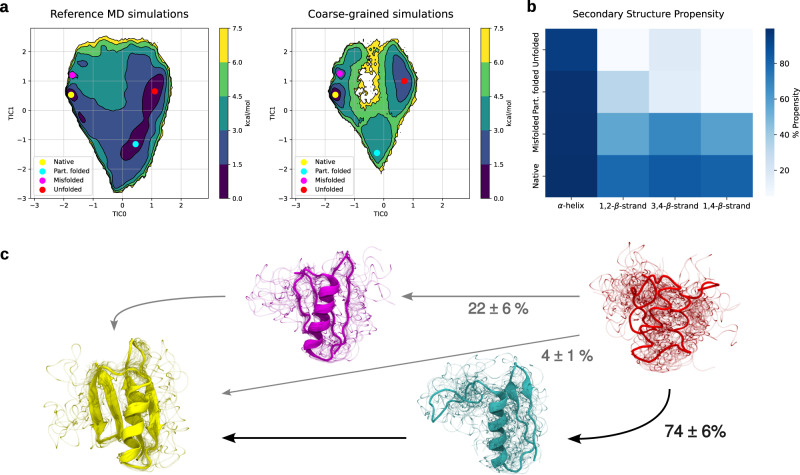
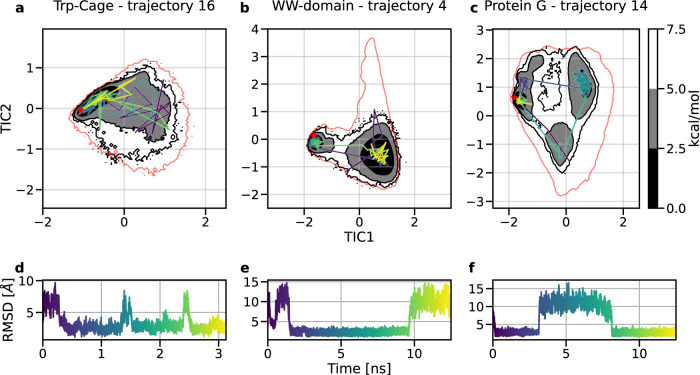
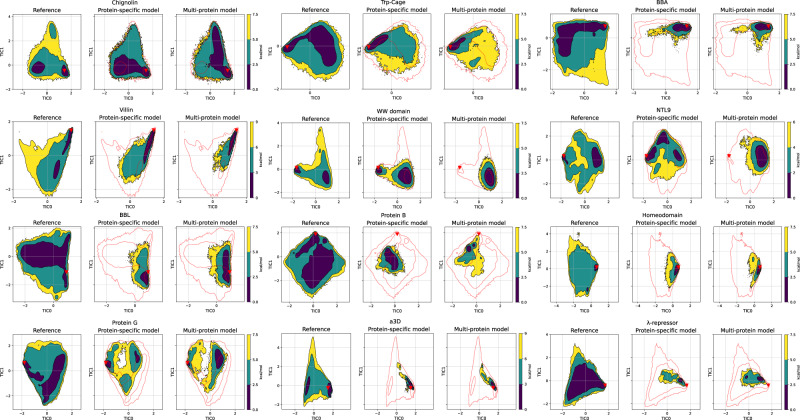
A generalized understanding of protein dynamics is an unsolved scientific problem, the solution of which is critical to the interpretation of the structure-function relationships that govern essential biological processes. Here, we approach this problem by constructing coarse-grained molecular potentials based on artificial neural networks and grounded in statistical mechanics. For training, we build a unique dataset of unbiased all-atom molecular dynamics simulations of approximately 9 ms for twelve different proteins with multiple secondary structure arrangements. The coarse-grained models are capable of accelerating the dynamics by more than three orders of magnitude while preserving the thermodynamics of the systems. Coarse-grained simulations identify relevant structural states in the ensemble with comparable energetics to the all-atom systems. Furthermore, we show that a single coarse-grained potential can integrate all twelve proteins and can capture experimental structural features of mutated proteins. These results indicate that machine learning coarse-grained potentials could provide a feasible approach to simulate and understand protein dynamics.
蛋白质动力学的普遍理解是一个未解决的科学问题,其解决方案对解释控制基本生物过程的结构-功能关系至关重要。在这里,我们通过构建基于人工神经网络和基于统计力学的粗粒度分子势来解决这个问题。在训练中,我们构建了一个独特的数据集,包含大约 9 毫秒的 12 种不同蛋白质的无偏全原子分子动力学模拟,这些蛋白质具有多种二级结构排列。粗粒度模型能够将动力学加速三个数量级以上,同时保持系统的热力学性质。粗粒度模拟可以在与全原子系统相当的能量水平上识别组合中的相关结构状态。此外,我们还表明,单个粗粒度势可以整合所有 12 种蛋白质,并可以捕获突变蛋白质的实验结构特征。这些结果表明,机器学习粗粒度势可以为模拟和理解蛋白质动力学提供一种可行的方法。