Professorship of Multiscale Modeling of Fluid Materials, TUM School of Engineering and Design, Technical University of Munich, Munich, Germany.
Munich Data Science Institute, Technical University of Munich, Munich, Germany.
Nat Commun. 2021 Nov 25;12(1):6884. doi: 10.1038/s41467-021-27241-4.
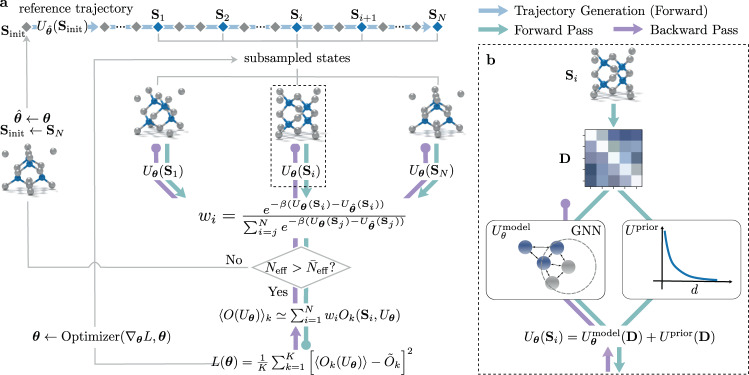
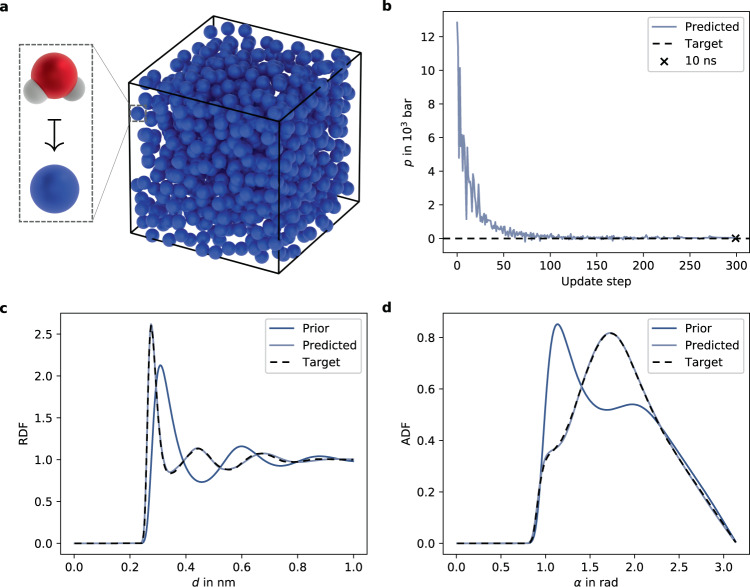
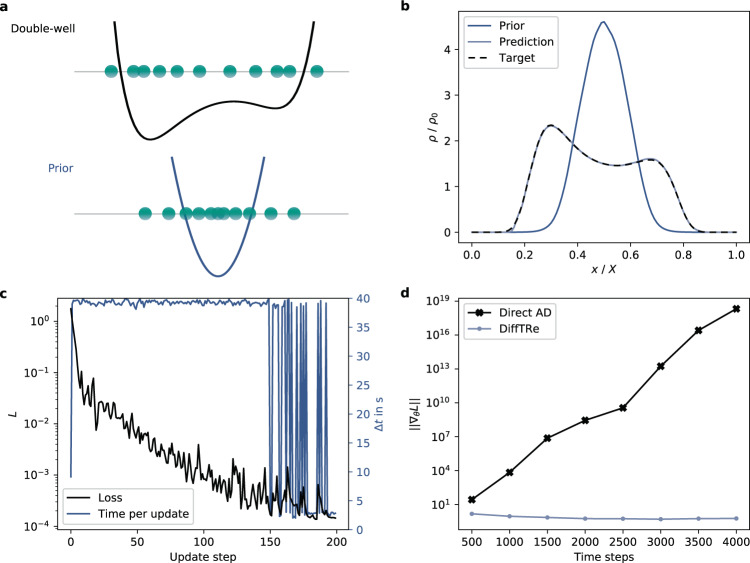
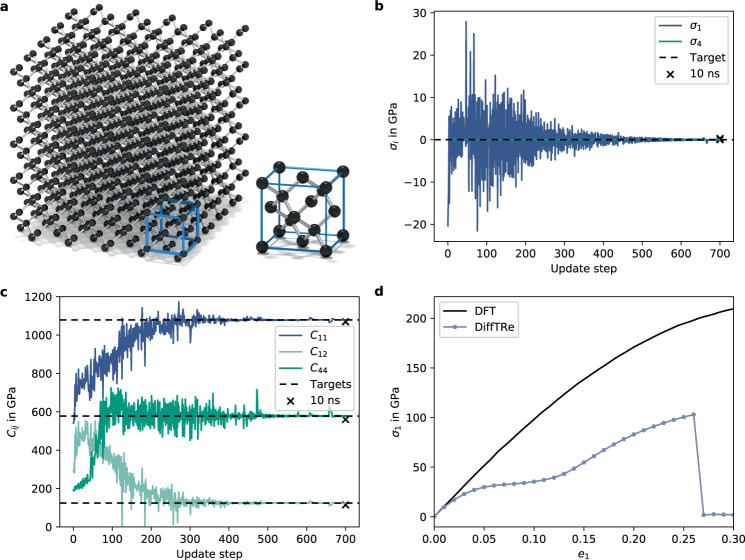
In molecular dynamics (MD), neural network (NN) potentials trained bottom-up on quantum mechanical data have seen tremendous success recently. Top-down approaches that learn NN potentials directly from experimental data have received less attention, typically facing numerical and computational challenges when backpropagating through MD simulations. We present the Differentiable Trajectory Reweighting (DiffTRe) method, which bypasses differentiation through the MD simulation for time-independent observables. Leveraging thermodynamic perturbation theory, we avoid exploding gradients and achieve around 2 orders of magnitude speed-up in gradient computation for top-down learning. We show effectiveness of DiffTRe in learning NN potentials for an atomistic model of diamond and a coarse-grained model of water based on diverse experimental observables including thermodynamic, structural and mechanical properties. Importantly, DiffTRe also generalizes bottom-up structural coarse-graining methods such as iterative Boltzmann inversion to arbitrary potentials. The presented method constitutes an important milestone towards enriching NN potentials with experimental data, particularly when accurate bottom-up data is unavailable.
在分子动力学 (MD) 中,最近基于量子力学数据自上而下训练的神经网络 (NN) 势取得了巨大的成功。而直接从实验数据中学习 NN 势的自顶向下方法则受到较少关注,当通过 MD 模拟反向传播时,通常会面临数值和计算方面的挑战。我们提出了可微分轨迹重加权 (DiffTRe) 方法,该方法绕过了 MD 模拟中对时间独立观测值的微分。利用热力学微扰理论,我们避免了梯度爆炸,并实现了自上而下学习中梯度计算的约 2 个数量级的加速。我们展示了 DiffTRe 在学习基于不同实验观测值(包括热力学、结构和力学性质)的金刚石原子模型和水的粗粒化模型的 NN 势方面的有效性。重要的是,DiffTRe 还将迭代 Boltzmann 反演等自下而上的结构粗粒化方法推广到任意势。所提出的方法是通过实验数据丰富 NN 势的一个重要里程碑,特别是在无法获得准确的自下而上数据时。