Department of Pharmacology and Toxicology, College of Medicine, University of Arkansas for Medical Sciences, Little Rock, AR, USA.
Department of Clinical Pharmacy, Faculty of Pharmacy, Cairo University, Cairo, Egypt.
Cell Death Dis. 2023 Sep 21;14(9):621. doi: 10.1038/s41419-023-06147-7.
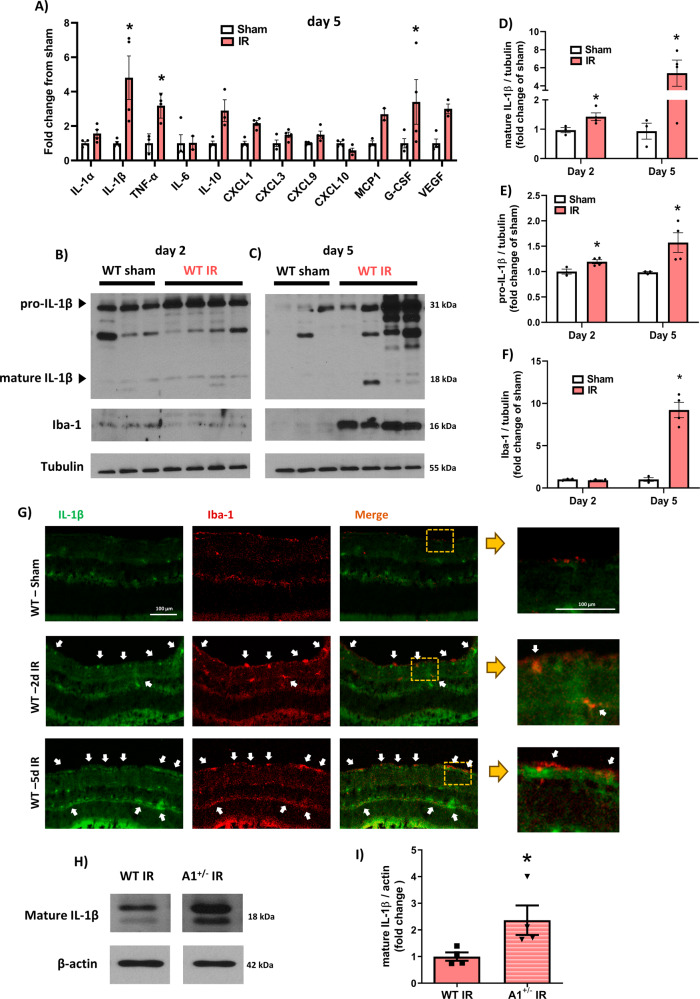
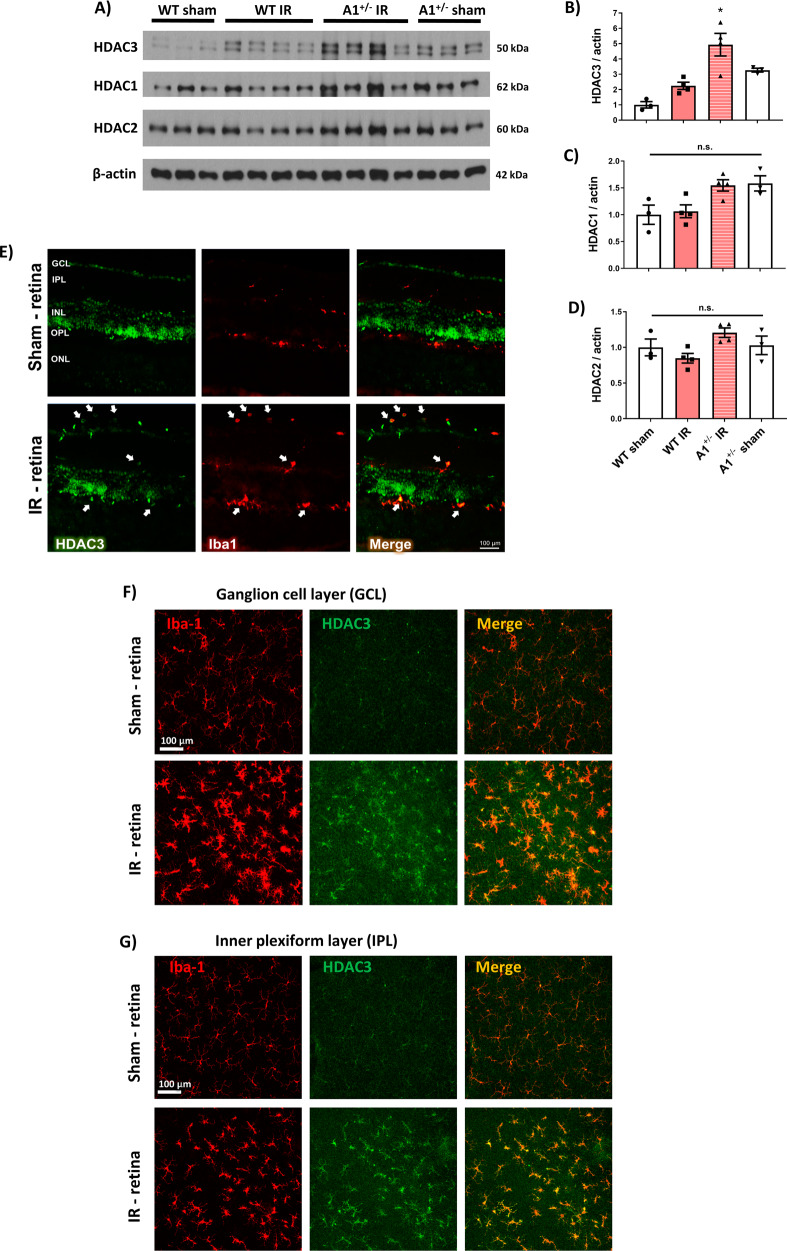
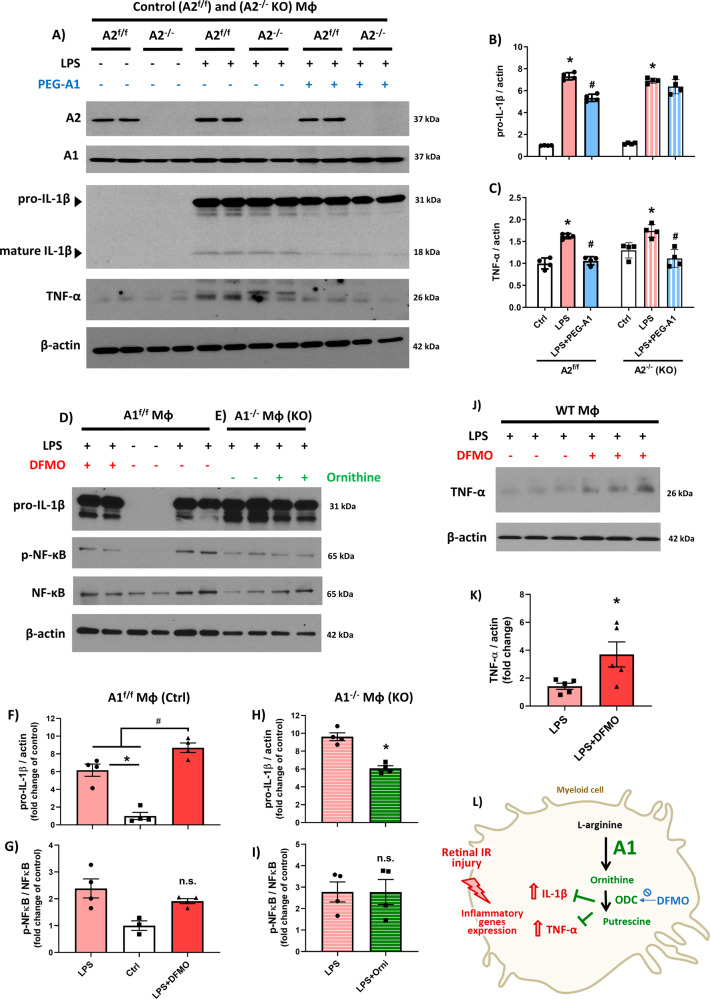
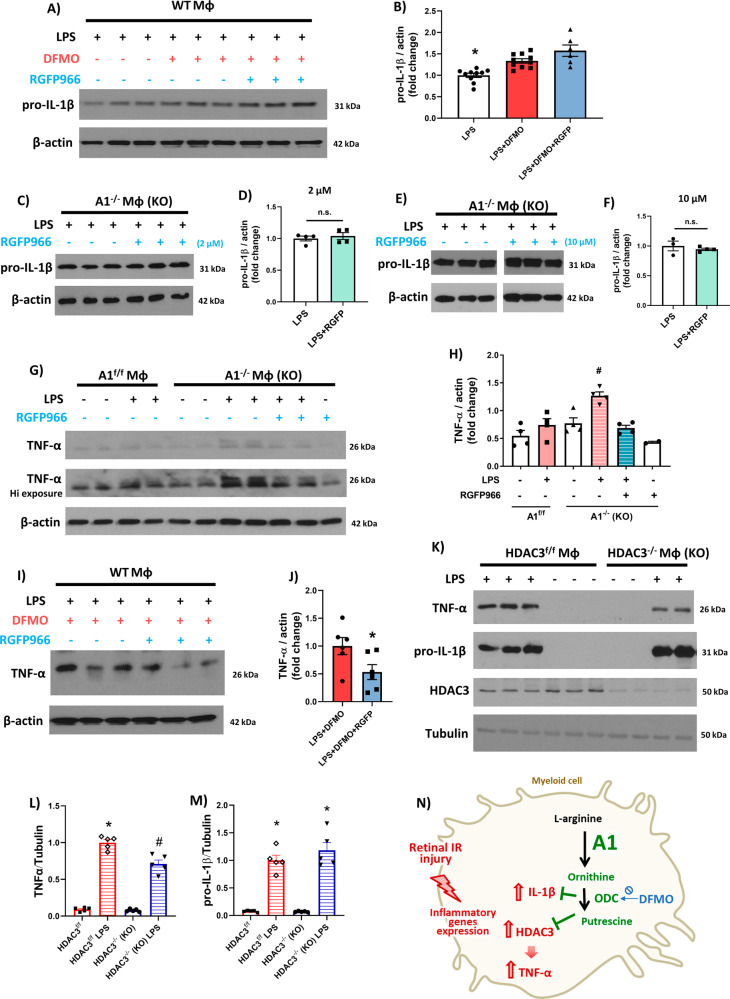
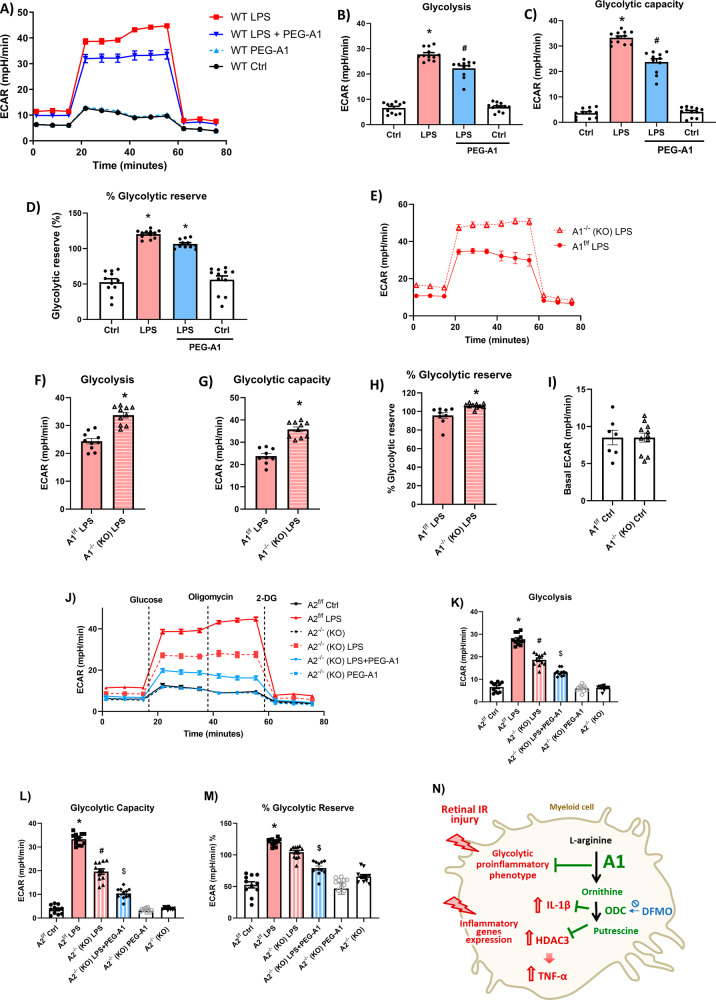
The enzyme arginase 1 (A1) hydrolyzes the amino acid arginine to form L-ornithine and urea. Ornithine is further converted to polyamines by the ornithine decarboxylase (ODC) enzyme. We previously reported that deletion of myeloid A1 in mice exacerbates retinal damage after ischemia/reperfusion (IR) injury. Furthermore, treatment with A1 protects against retinal IR injury in wild-type mice. PEG-A1 also mitigates the exaggerated inflammatory response of A1 knockout (KO) macrophages in vitro. Here, we sought to identify the anti-inflammatory pathway that confers macrophage A1-mediated protection against retinal IR injury. Acute elevation of intraocular pressure was used to induce retinal IR injury in mice. A multiplex cytokine assay revealed a marked increase in the inflammatory cytokines interleukin 1β (IL-1β) and tumor necrosis factor α (TNF-α) in the retina at day 5 after IR injury. In vitro, blocking the A1/ODC pathway augmented IL-1β and TNF-α production in stimulated macrophages. Furthermore, A1 treatment attenuated the stimulated macrophage metabolic switch to a pro-inflammatory glycolytic phenotype, whereas A1 deletion had the opposite effect. Screening for histone deacetylases (HDACs) which play a role in macrophage inflammatory response showed that A1 deletion or ODC inhibition increased the expression of HDAC3. We further showed the involvement of HDAC3 in the upregulation of TNF-α but not IL-1β in stimulated macrophages deficient in the A1/ODC pathway. Investigating HDAC3 KO macrophages showed a reduced inflammatory response and a less glycolytic phenotype upon stimulation. In vivo, HDAC3 co-localized with microglia/macrophages at day 2 after IR in WT retinas and was further increased in A1-deficient retinas. Collectively, our data provide initial evidence that A1 exerts its anti-inflammatory effect in macrophages via ODC-mediated suppression of HDAC3 and IL-1β. Collectively we propose that interventions that augment the A1/ODC pathway and inhibit HDAC3 may confer therapeutic benefits for the treatment of retinal ischemic diseases.
酶精氨酸酶 1(A1)将氨基酸精氨酸水解形成 L-鸟氨酸和尿素。鸟氨酸进一步被鸟氨酸脱羧酶(ODC)酶转化为多胺。我们之前报道过,在小鼠中缺失髓样 A1 会加剧缺血/再灌注(IR)损伤后的视网膜损伤。此外,在野生型小鼠中,A1 的治疗可保护视网膜免受 IR 损伤。PEG-A1 还可减轻 A1 敲除(KO)巨噬细胞在体外的过度炎症反应。在这里,我们试图确定赋予巨噬细胞 A1 介导的对视网膜 IR 损伤的保护作用的抗炎途径。急性升高眼内压用于诱导小鼠视网膜 IR 损伤。多重细胞因子分析显示,IR 损伤后 5 天,视网膜中炎症细胞因子白细胞介素 1β(IL-1β)和肿瘤坏死因子α(TNF-α)明显增加。在体外,阻断 A1/ODC 途径会增加刺激的巨噬细胞中 IL-1β 和 TNF-α 的产生。此外,A1 处理可减弱刺激的巨噬细胞向促炎糖酵解表型的代谢转变,而 A1 缺失则有相反的作用。筛选在巨噬细胞炎症反应中起作用的组蛋白去乙酰化酶(HDACs)显示,A1 缺失或 ODC 抑制增加了 HDAC3 的表达。我们进一步表明,HDAC3 参与了刺激的 A1/ODC 途径缺陷的巨噬细胞中 TNF-α的上调,但不参与 IL-1β 的上调。研究 HDAC3 KO 巨噬细胞显示,在刺激下炎症反应减少,糖酵解表型减少。在体内,在 WT 视网膜中,IR 后 2 天 HDAC3 与小胶质细胞/巨噬细胞共定位,并在 A1 缺陷型视网膜中进一步增加。总之,我们的数据提供了初步证据,表明 A1 通过 ODC 介导的 HDAC3 和 IL-1β 抑制在巨噬细胞中发挥抗炎作用。总的来说,我们提出增强 A1/ODC 途径和抑制 HDAC3 的干预措施可能为治疗视网膜缺血性疾病提供治疗益处。