Mani Hemalatha, Chang Chun-Chun, Hsu Hao-Jen, Yang Chin-Hao, Yen Jui-Hung, Liou Je-Wen
Institute of Medical Sciences, Tzu Chi University, Hualien 97004, Taiwan.
Department of Laboratory Medicine, Hualien Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, Hualien 97004, Taiwan.
Bioengineering (Basel). 2023 Aug 24;10(9):1004. doi: 10.3390/bioengineering10091004.
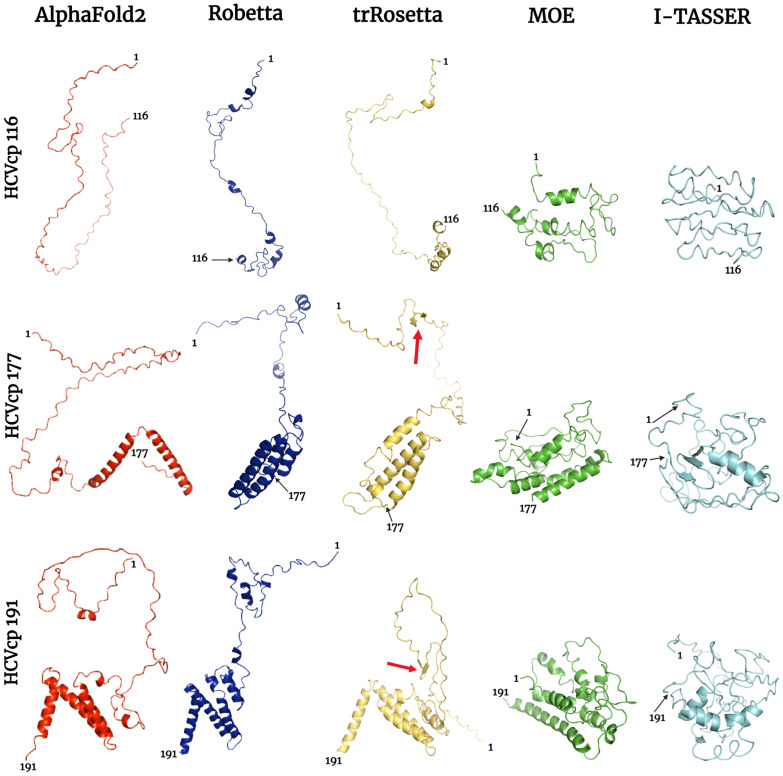
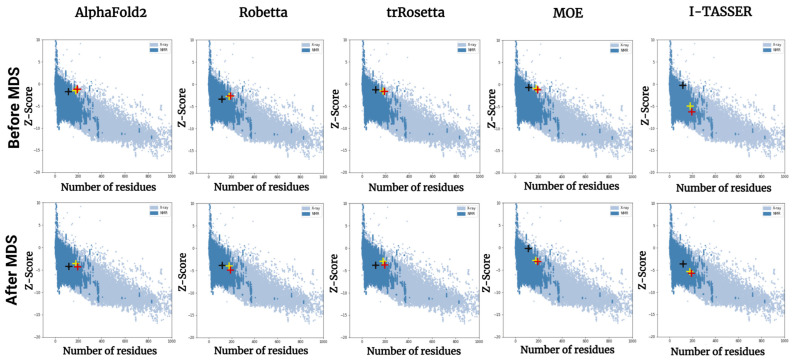
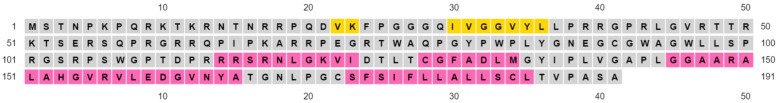
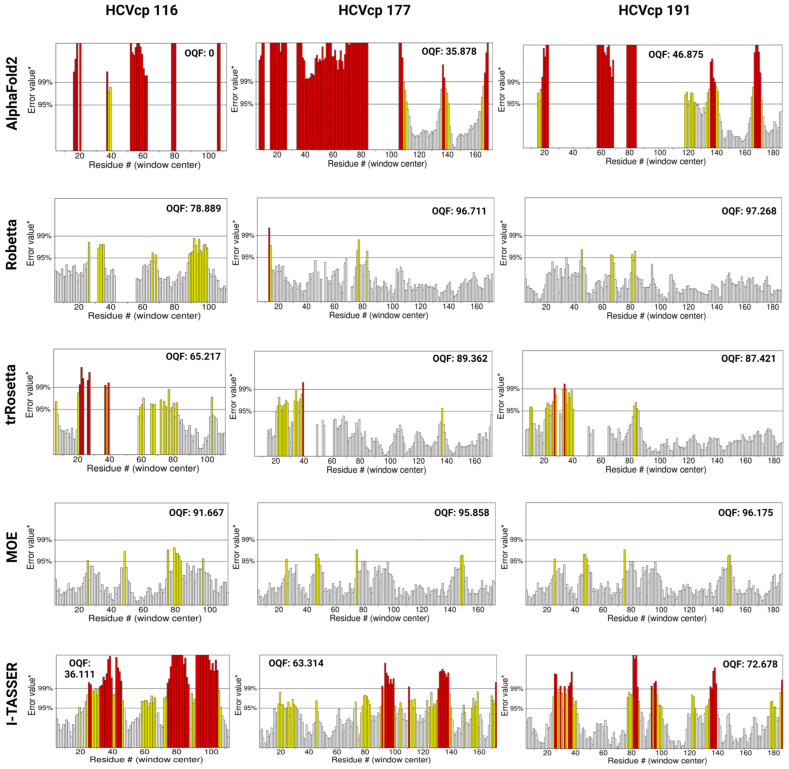
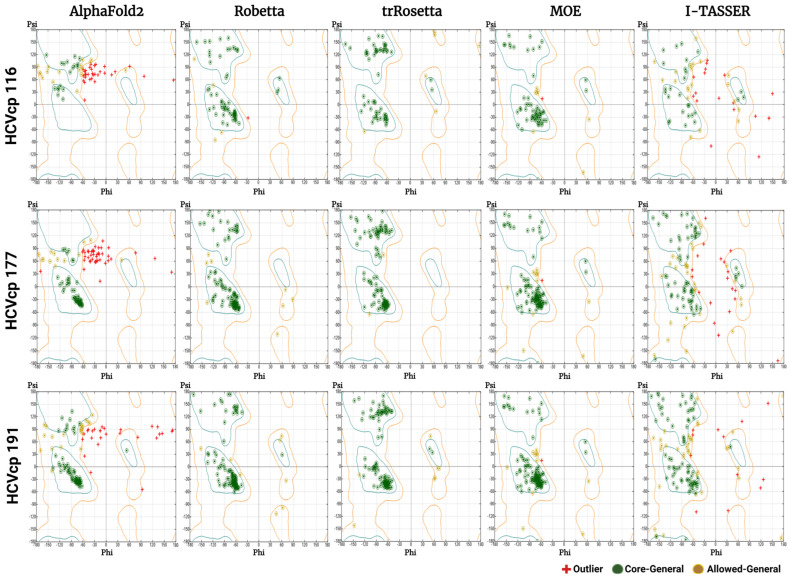
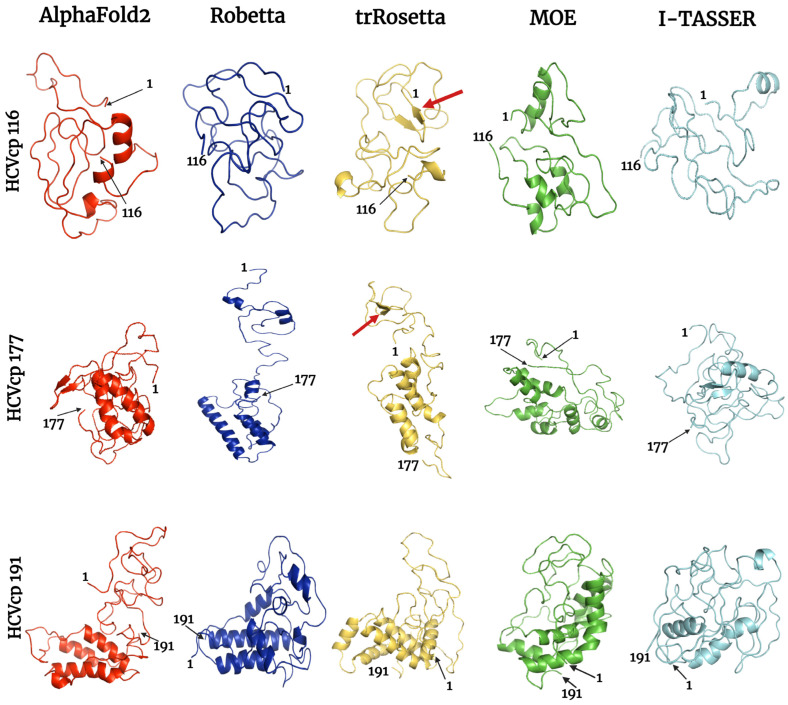
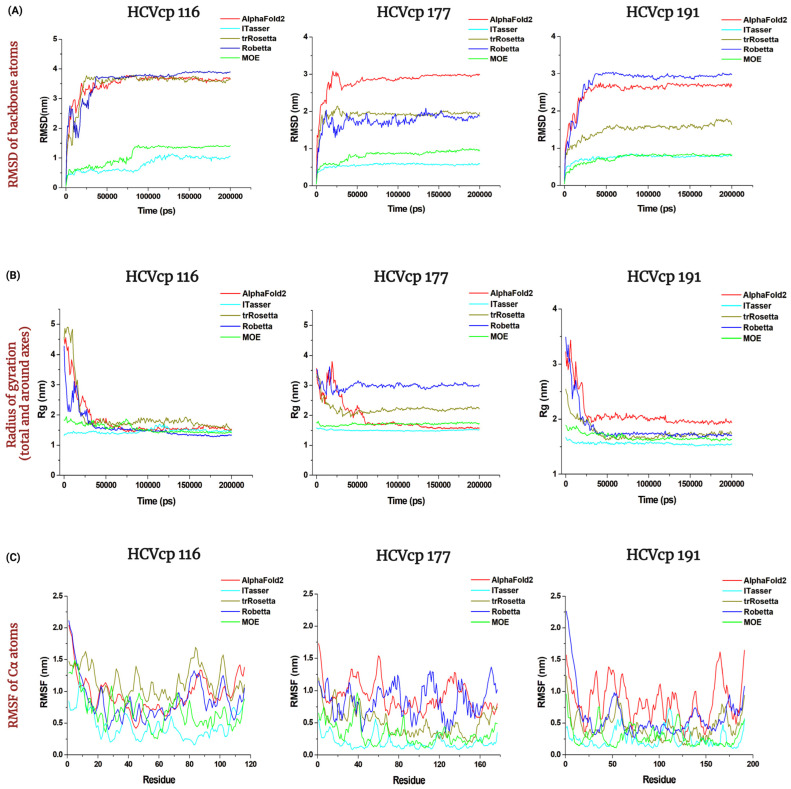
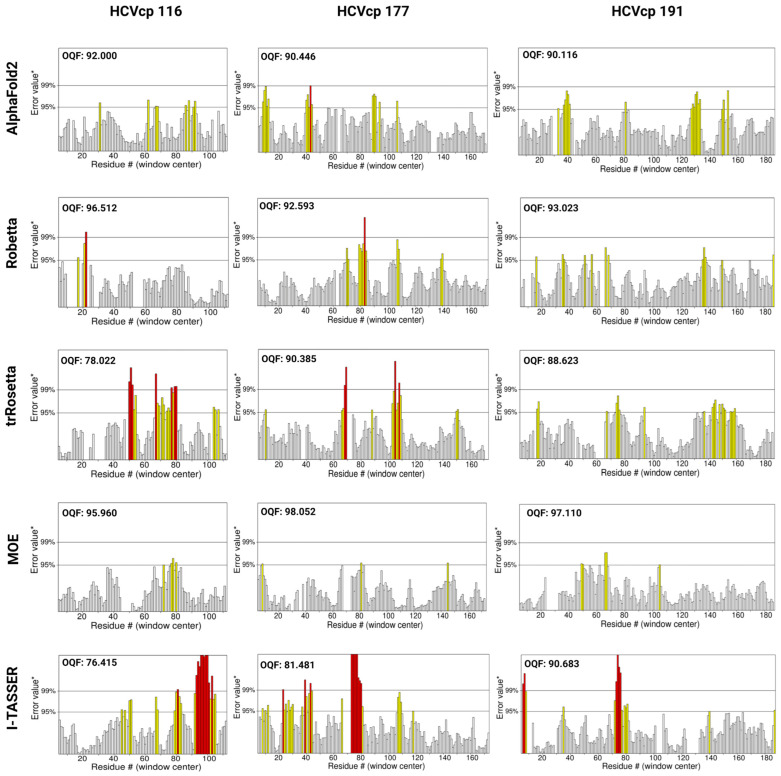
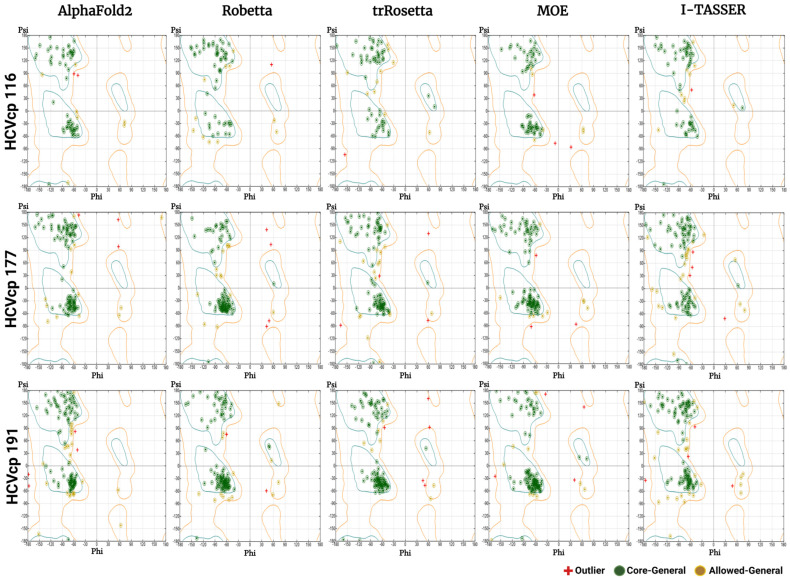
The structural analysis of proteins is a major domain of biomedical research. Such analysis requires resolved three-dimensional structures of proteins. Advancements in computer technology have led to progress in biomedical research. In silico prediction and modeling approaches have facilitated the construction of protein structures, with or without structural templates. In this study, we used three neural network-based de novo modeling approaches-AlphaFold2 (AF2), Robetta-RoseTTAFold (Robetta), and transform-restrained Rosetta (trRosetta)-and two template-based tools-the Molecular Operating Environment (MOE) and iterative threading assembly refinement (I-TASSER)-to construct the structure of a viral capsid protein, hepatitis C virus core protein (HCVcp), whose structure have not been fully resolved by laboratory techniques. Templates with sufficient sequence identity for the homology modeling of complete HCVcp are currently unavailable. Therefore, we performed domain-based homology modeling for MOE simulations. The templates for each domain were obtained through sequence-based searches on NCBI and the Protein Data Bank. Then, the modeled domains were assembled to construct the complete structure of HCVcp. The full-length structure and two truncated forms modeled using various computational tools were compared. Molecular dynamics (MD) simulations were performed to refine the structures. The root mean square deviation of backbone atoms, root mean square fluctuation of Cα atoms, and radius of gyration were calculated to monitor structural changes and convergence in the simulations. The model quality was evaluated through ERRAT and phi-psi plot analysis. In terms of the initial prediction for protein modeling, Robetta and trRosetta outperformed AF2. Regarding template-based tools, MOE outperformed I-TASSER. MD simulations resulted in compactly folded protein structures, which were of good quality and theoretically accurate. Thus, the predicted structures of certain proteins must be refined to obtain reliable structural models. MD simulation is a promising tool for this purpose.
蛋白质的结构分析是生物医学研究的一个主要领域。这种分析需要蛋白质的解析三维结构。计算机技术的进步推动了生物医学研究的发展。计算机模拟预测和建模方法促进了蛋白质结构的构建,无论有无结构模板。在本研究中,我们使用了三种基于神经网络的从头建模方法——AlphaFold2(AF2)、Robetta-RoseTTAFold(Robetta)和变换约束Rosetta(trRosetta)——以及两种基于模板的工具——分子操作环境(MOE)和迭代穿线装配优化(I-TASSER)——来构建一种病毒衣壳蛋白丙型肝炎病毒核心蛋白(HCVcp)的结构,其实验室技术尚未完全解析其结构。目前尚无具有足够序列同一性用于完整HCVcp同源建模的模板。因此,我们对MOE模拟进行了基于结构域的同源建模。每个结构域的模板通过在NCBI和蛋白质数据库上基于序列的搜索获得。然后,将建模的结构域组装起来构建HCVcp的完整结构。比较了使用各种计算工具建模的全长结构和两种截短形式。进行分子动力学(MD)模拟以优化结构。计算主链原子的均方根偏差、Cα原子的均方根波动和回转半径,以监测模拟中的结构变化和收敛情况。通过ERRAT和phi-psi图分析评估模型质量。在蛋白质建模的初始预测方面,Robetta和trRosetta优于AF2。在基于模板的工具方面,MOE优于I-TASSER。MD模拟产生了紧密折叠的蛋白质结构,质量良好且理论上准确。因此,必须对某些蛋白质的预测结构进行优化以获得可靠的结构模型。MD模拟是实现这一目的的一种有前途的工具。