Division of Infectious Diseases and Division of Computer Aided Drug Design, The Red-Green Research Centre, BICCB, Tejgaon, Dhaka, Bangladesh.
Department of Physical Sciences, University of Arkansas - Fort Smith, Fort Smith, AR, USA.
J Biomol Struct Dyn. 2022 Jul;40(10):4725-4738. doi: 10.1080/07391102.2020.1861983. Epub 2020 Dec 22.
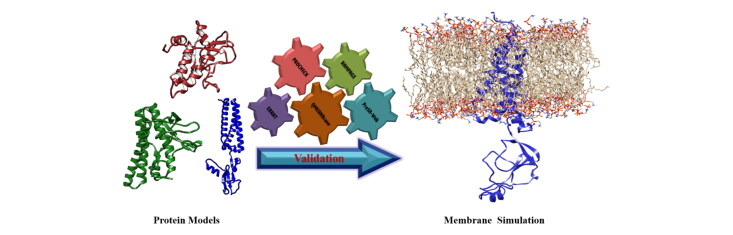
SARS-CoV-2 membrane (M) protein performs a variety of critical functions in virus infection cycle. However, the expression and purification of membrane protein structure is difficult despite tremendous progress. In this study, the 3 D structure is modeled followed by intensive validation and molecular dynamics simulation. The lack of suitable homologous templates (>30% sequence identities) leads us to construct the membrane protein models using template-free modeling ( or ) approach with Robetta and trRosetta servers. Comparing with other model structures, it is evident that trRosetta (TM-score: 0.64; TM region RMSD: 2 Å) can provide the best model than Robetta (TM-score: 0.61; TM region RMSD: 3.3 Å) and I-TASSER (TM-score: 0.45; TM region RMSD: 6.5 Å). 100 ns molecular dynamics simulations are performed on the model structures by incorporating membrane environment. Moreover, secondary structure elements and principal component analysis (PCA) have also been performed on MD simulation data. Finally, trRosetta model is utilized for interpretation and visualization of interacting residues during protein-protein interactions. The common interacting residues including Phe103, Arg107, Met109, Trp110, Arg131, and Glu135 in the C-terminal domain of M protein are identified in membrane-spike and membrane-nucleocapsid protein complexes. The active site residues are also predicted for potential drug and peptide binding. Overall, this study might be helpful to design drugs and peptides against the modeled membrane protein of SARS-CoV-2 to accelerate further investigation. Communicated by Ramaswamy H. Sarma.
SARS-CoV-2 膜(M)蛋白在病毒感染周期中发挥多种关键功能。然而,尽管取得了巨大的进展,膜蛋白结构的表达和纯化仍然很困难。在本研究中,对 3D 结构进行建模,然后进行了密集的验证和分子动力学模拟。由于缺乏合适的同源模板(>30%的序列同一性),我们使用无模板建模(或)方法使用 Robetta 和 trRosetta 服务器构建膜蛋白模型。与其他模型结构相比,trRosetta(TM-score:0.64;TM 区域 RMSD:2 Å)可以提供比 Robetta(TM-score:0.61;TM 区域 RMSD:3.3 Å)和 I-TASSER(TM-score:0.45;TM 区域 RMSD:6.5 Å)更好的模型。通过纳入膜环境,对模型结构进行了 100 ns 的分子动力学模拟。此外,还对 MD 模拟数据进行了二级结构元素和主成分分析(PCA)。最后,利用 trRosetta 模型对膜蛋白相互作用模型中相互作用残基进行解释和可视化。在膜刺突和膜核衣壳蛋白复合物中鉴定到 M 蛋白 C 末端结构域中常见的相互作用残基包括 Phe103、Arg107、Met109、Trp110、Arg131 和 Glu135。还预测了活性位点残基,以寻找潜在的药物和肽结合物。总的来说,这项研究可能有助于设计针对 SARS-CoV-2 膜蛋白的药物和肽,以加速进一步的研究。由 Ramaswamy H. Sarma 传达。