Vollger Mitchell R, Korlach Jonas, Eldred Kiara C, Swanson Elliott, Underwood Jason G, Cheng Yong-Han H, Ranchalis Jane, Mao Yizi, Blue Elizabeth E, Schwarze Ulrike, Munson Katherine M, Saunders Christopher T, Wenger Aaron M, Allworth Aimee, Chanprasert Sirisak, Duerden Brittney L, Glass Ian, Horike-Pyne Martha, Kim Michelle, Leppig Kathleen A, McLaughlin Ian J, Ogawa Jessica, Rosenthal Elisabeth A, Sheppeard Sam, Sherman Stephanie M, Strohbehn Samuel, Yuen Amy L, Reh Thomas A, Byers Peter H, Bamshad Michael J, Hisama Fuki M, Jarvik Gail P, Sancak Yasemin, Dipple Katrina M, Stergachis Andrew B
University of Washington School of Medicine, Department of Genome Sciences, Seattle, WA, USA.
University of Washington School of Medicine, Department of Medicine, Seattle, WA, USA.
bioRxiv. 2023 Sep 27:2023.09.26.559521. doi: 10.1101/2023.09.26.559521.
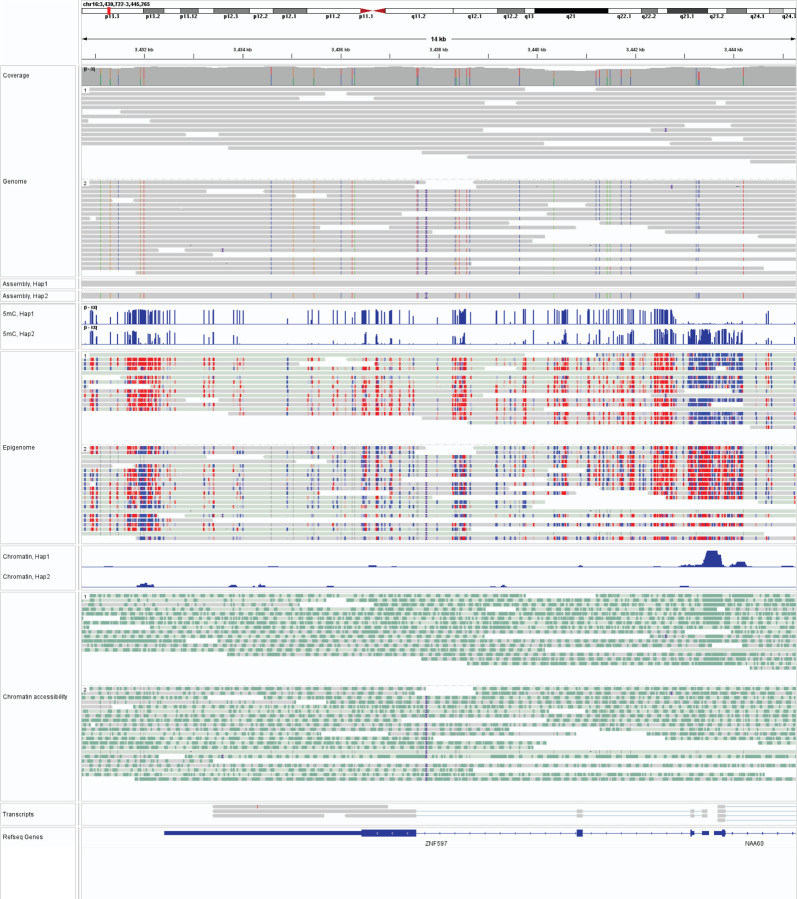
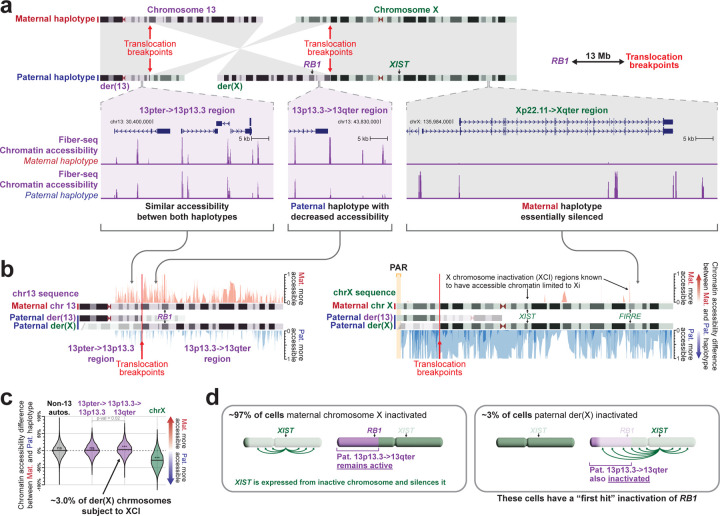
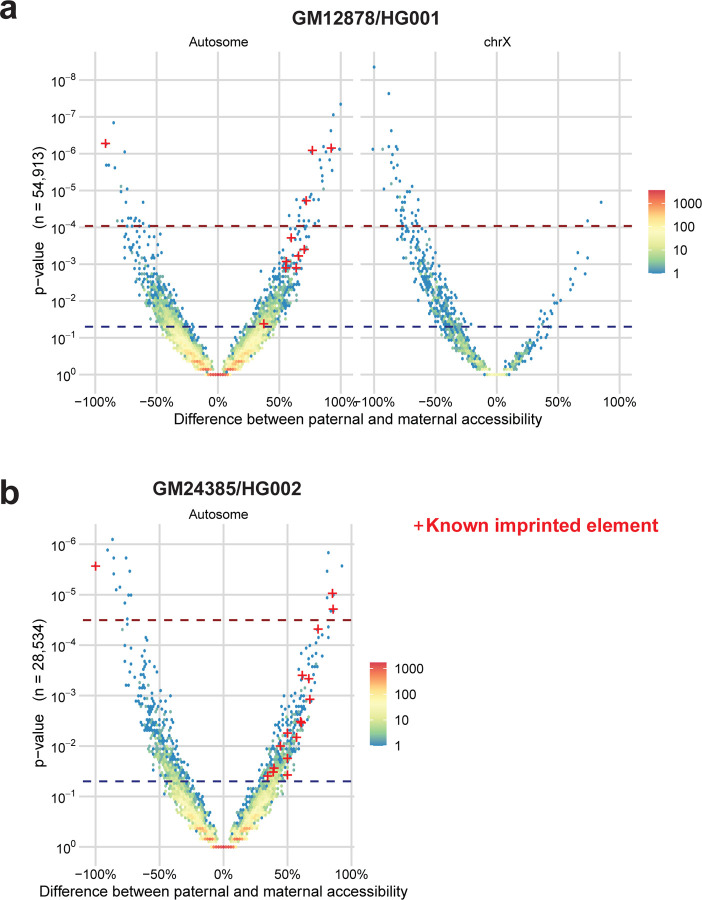
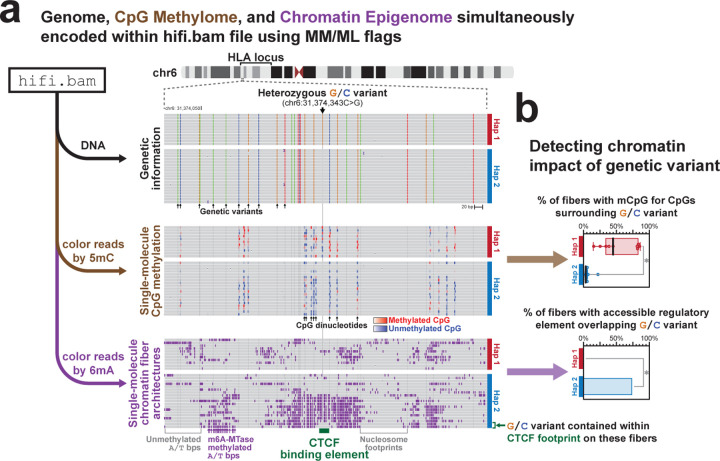
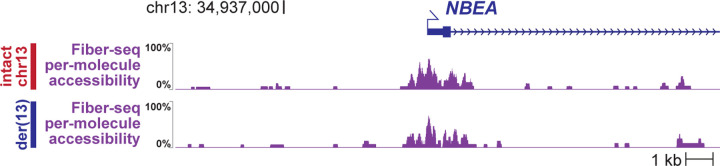
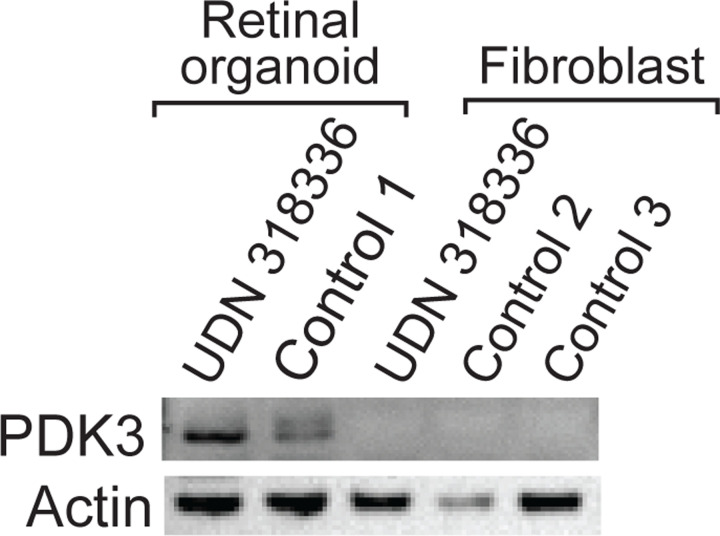
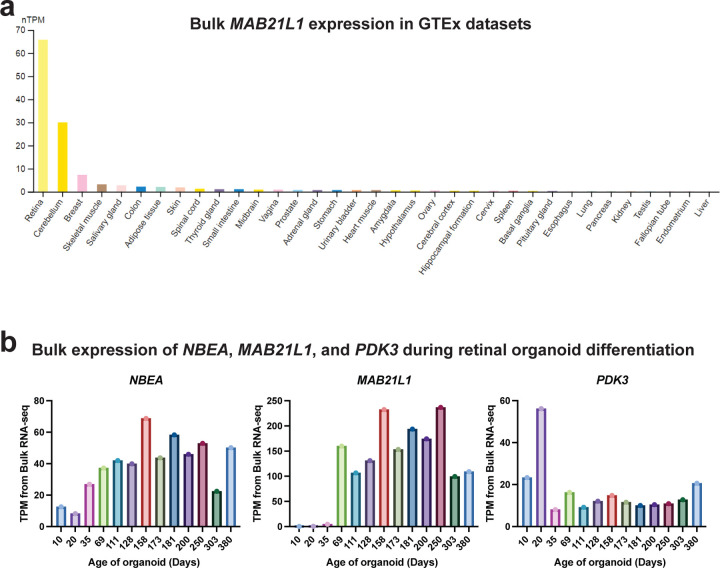
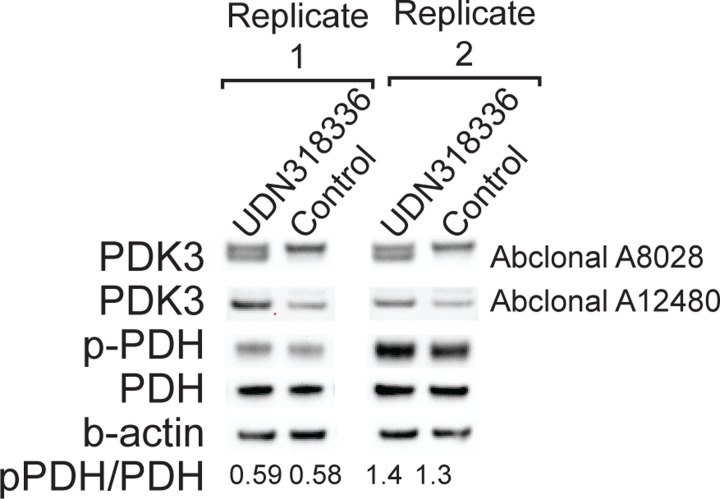
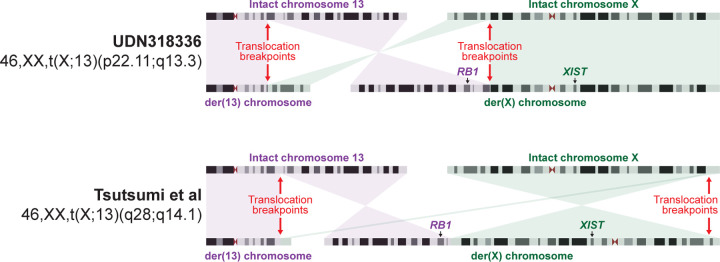
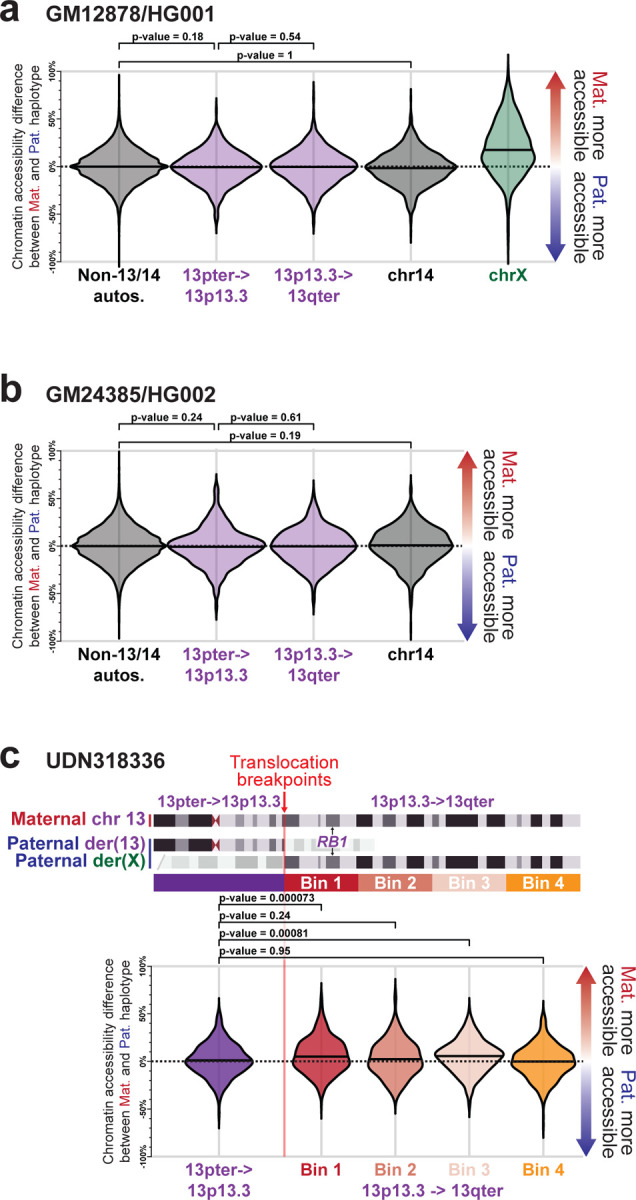
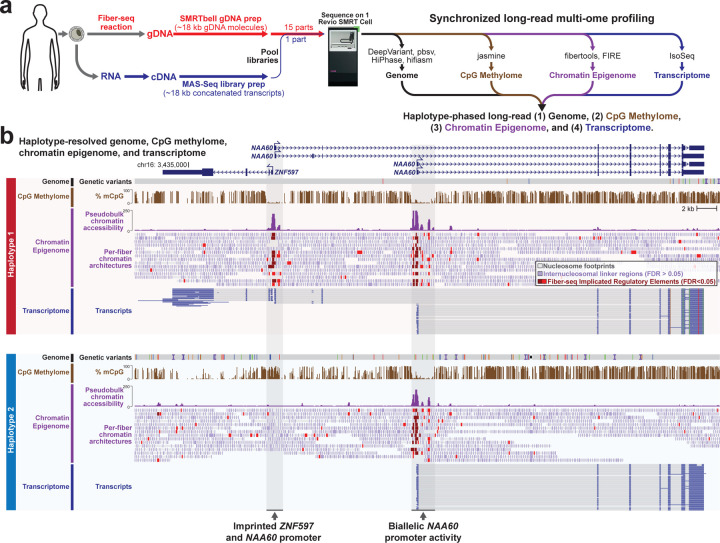
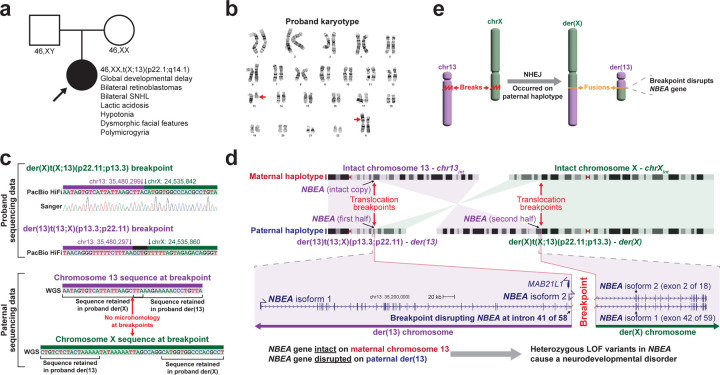
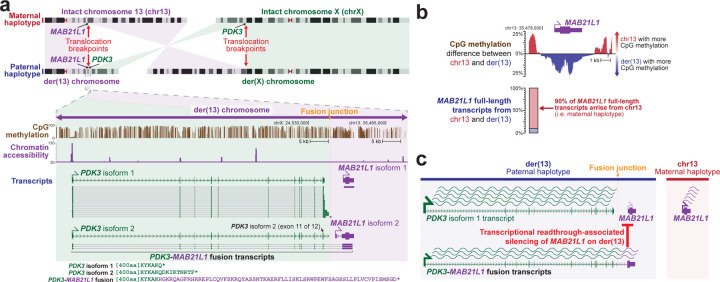
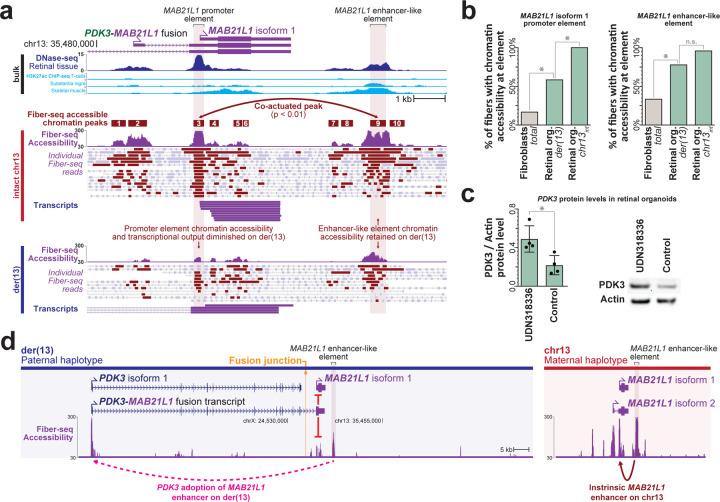
Resolving the molecular basis of a Mendelian condition (MC) remains challenging owing to the diverse mechanisms by which genetic variants cause disease. To address this, we developed a synchronized long-read genome, methylome, epigenome, and transcriptome sequencing approach, which enables accurate single-nucleotide, insertion-deletion, and structural variant calling and diploid genome assembly, and permits the simultaneous elucidation of haplotype-resolved CpG methylation, chromatin accessibility, and full-length transcript information in a single long-read sequencing run. Application of this approach to an Undiagnosed Diseases Network (UDN) participant with a chromosome X;13 balanced translocation of uncertain significance revealed that this translocation disrupted the functioning of four separate genes (, , , and ) previously associated with single-gene MCs. Notably, the function of each gene was disrupted via a distinct mechanism that required integration of the four 'omes' to resolve. These included nonsense-mediated decay, fusion transcript formation, enhancer adoption, transcriptional readthrough silencing, and inappropriate X chromosome inactivation of autosomal genes. Overall, this highlights the utility of synchronized long-read multi-omic profiling for mechanistically resolving complex phenotypes.
由于遗传变异导致疾病的机制多种多样,解析孟德尔疾病(MC)的分子基础仍然具有挑战性。为了解决这一问题,我们开发了一种同步长读长基因组、甲基化组、表观基因组和转录组测序方法,该方法能够进行准确的单核苷酸、插入缺失和结构变异检测以及二倍体基因组组装,并允许在一次长读长测序运行中同时阐明单倍型解析的CpG甲基化、染色质可及性和全长转录本信息。将该方法应用于未确诊疾病网络(UDN)中一名具有意义不明的X染色体;13号染色体平衡易位的参与者,结果显示该易位破坏了四个先前与单基因MC相关的独立基因(、、和)的功能。值得注意的是,每个基因的功能通过一种独特的机制被破坏,这种机制需要整合四个“组学”来解析。这些机制包括无义介导的衰变、融合转录本形成、增强子采用、转录通读沉默以及常染色体基因不适当的X染色体失活。总体而言,这突出了同步长读长多组学分析在机械解析复杂表型方面的实用性。