Lam Tanya, Rocca Clarissa, Ibanez Kristina, Dalmia Anupriya, Tallman Samuel, Hadjivassiliou Marios, Hensiek Anke, Nemeth Andrea, Facchini Stefano, Wood Nicholas, Cortese Andrea, Houlden Henry, Tucci Arianna
Department of Clinical Genetics, Great Ormond Street Hospital NHS Trust, London, WC1N 3JH, UK.
Clinical Pharmacology, William Harvey Research Institute, School of Medicine and Dentistry, Queen Mary University of London, London, EC1M 6BQ, UK.
Brain Commun. 2023 Sep 14;5(5):fcad244. doi: 10.1093/braincomms/fcad244. eCollection 2023.
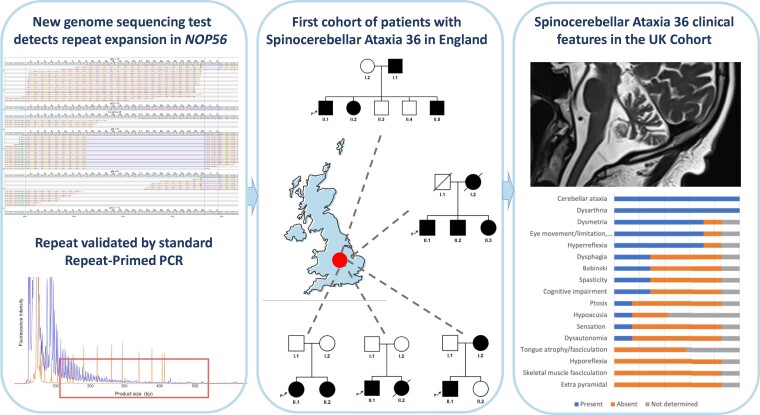
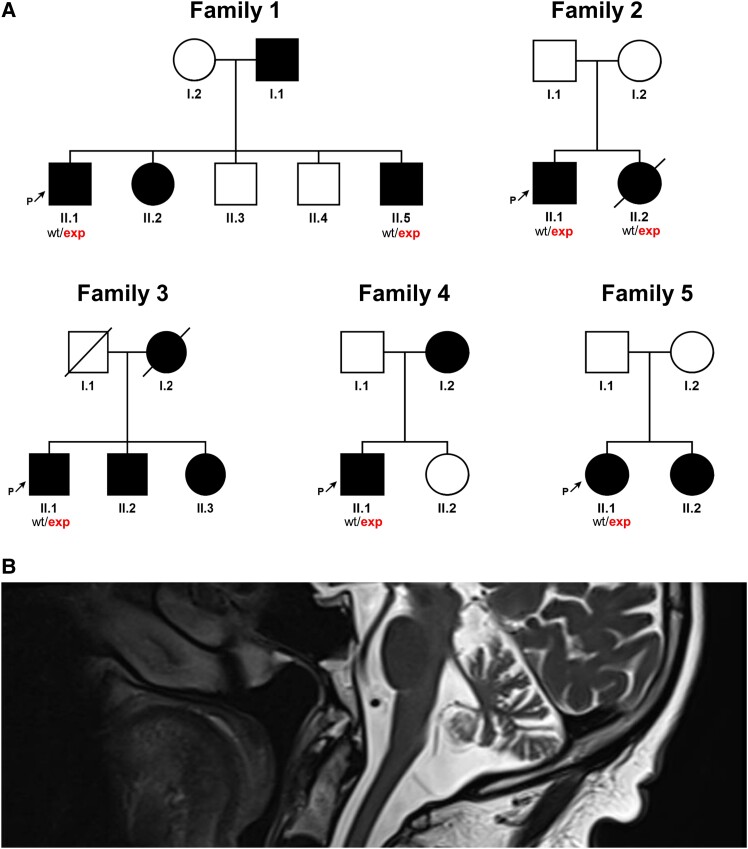
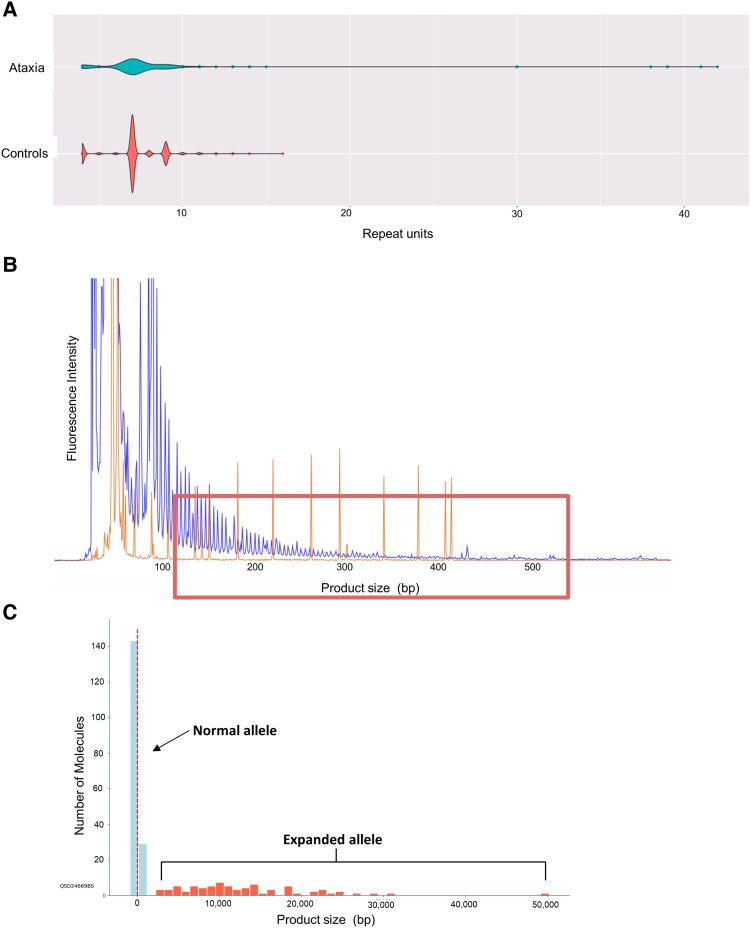
Spinocerebellar ataxias form a clinically and genetically heterogeneous group of neurodegenerative disorders characterized by progressive cerebellar ataxia. Their prevalence varies among populations and ethnicities. Spinocerebellar ataxia 36 is caused by a GGCCTG repeat expansion in the first intron of the gene and is characterized by late-onset ataxia, sensorineural hearing loss and upper and lower motor neuron signs, including tongue fasciculations. Spinocerebellar ataxia 36 has been described mainly in East Asian and Western European patients and was thought to be absent in the British population. Leveraging novel bioinformatic tools to detect repeat expansions from whole-genome sequencing, we analyse the repeat in 1257 British patients with hereditary ataxia and in 7506 unrelated controls. We identify pathogenic repeat expansions in five families (seven patients), representing the first cohort of White British descent patients with spinocerebellar ataxia 36. Employing approaches using whole-genome sequencing data, we found an 87 kb shared haplotype in among the affected individuals from five families around the repeat region, although this block was also shared between several controls, suggesting that the repeat arises on a permissive haplotype. Clinically, the patients presented with slowly progressive cerebellar ataxia with a low rate of hearing loss and variable rates of motor neuron impairment. Our findings show that the expansion causes ataxia in the British population and that spinocerebellar ataxia 36 can be suspected in patients with a late-onset, slowly progressive ataxia, even without the findings of hearing loss and tongue fasciculation.
脊髓小脑共济失调是一组临床和遗传异质性的神经退行性疾病,其特征为进行性小脑共济失调。其患病率在不同人群和种族中有所不同。脊髓小脑共济失调36型由基因第一内含子中的GGCCTG重复序列扩增引起,其特征为迟发性共济失调、感音神经性听力损失以及上下运动神经元体征,包括舌肌束颤。脊髓小脑共济失调36型主要在东亚和西欧患者中被描述,此前认为在英国人群中不存在。利用新型生物信息学工具从全基因组测序中检测重复序列扩增,我们分析了1257例英国遗传性共济失调患者和7506例无关对照中的重复序列。我们在五个家系(7例患者)中鉴定出致病性重复序列扩增,这代表了首批有脊髓小脑共济失调36型的英国白人后裔患者队列。采用基于全基因组测序数据的方法,我们在五个家系的受影响个体中发现了围绕重复序列区域的一个87 kb共享单倍型,尽管该区域在几个对照中也有共享,这表明该重复序列出现在一个允许的单倍型上。临床上,这些患者表现为缓慢进展的小脑共济失调,听力损失发生率低,运动神经元损害发生率各异。我们的研究结果表明,该重复序列扩增在英国人群中可导致共济失调,并且对于迟发性、缓慢进展性共济失调患者即使没有听力损失和舌肌束颤的表现,也可怀疑为脊髓小脑共济失调36型。