Ibrahimi Eliana, Lopes Marta B, Dhamo Xhilda, Simeon Andrea, Shigdel Rajesh, Hron Karel, Stres Blaž, D'Elia Domenica, Berland Magali, Marcos-Zambrano Laura Judith
Department of Biology, Faculty of Natural Sciences, University of Tirana, Tirana, Albania.
Department of Mathematics, Center for Mathematics and Applications (NOVA Math), NOVA School of Science and Technology, Caparica, Portugal.
Front Microbiol. 2023 Oct 5;14:1250909. doi: 10.3389/fmicb.2023.1250909. eCollection 2023.
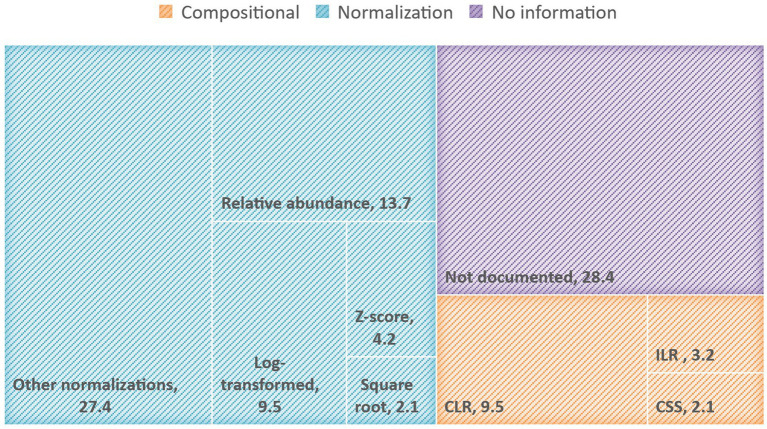
Although metagenomic sequencing is now the preferred technique to study microbiome-host interactions, analyzing and interpreting microbiome sequencing data presents challenges primarily attributed to the statistical specificities of the data (e.g., sparse, over-dispersed, compositional, inter-variable dependency). This mini review explores preprocessing and transformation methods applied in recent human microbiome studies to address microbiome data analysis challenges. Our results indicate a limited adoption of transformation methods targeting the statistical characteristics of microbiome sequencing data. Instead, there is a prevalent usage of relative and normalization-based transformations that do not specifically account for the specific attributes of microbiome data. The information on preprocessing and transformations applied to the data before analysis was incomplete or missing in many publications, leading to reproducibility concerns, comparability issues, and questionable results. We hope this mini review will provide researchers and newcomers to the field of human microbiome research with an up-to-date point of reference for various data transformation tools and assist them in choosing the most suitable transformation method based on their research questions, objectives, and data characteristics.
尽管宏基因组测序现在是研究微生物组与宿主相互作用的首选技术,但分析和解释微生物组测序数据存在挑战,这些挑战主要归因于数据的统计学特性(例如,稀疏、过度分散、组成性、变量间依赖性)。本综述探讨了近期人类微生物组研究中应用的预处理和转换方法,以应对微生物组数据分析挑战。我们的结果表明,针对微生物组测序数据统计特征的转换方法应用有限。相反,普遍使用的是基于相对和归一化的转换,这些转换并未特别考虑微生物组数据的特定属性。许多出版物中关于分析前应用于数据的预处理和转换的信息不完整或缺失,导致了可重复性问题、可比性问题以及结果存疑。我们希望本综述能为人类微生物组研究领域的研究人员和新手提供各种数据转换工具的最新参考点,并帮助他们根据研究问题、目标和数据特征选择最合适的转换方法。