Department of Mathematics and Statistics, University of Otago, Dunedin 9054, New Zealand
Invermay Agricultural Centre, AgResearch, Mosgiel 9053, New Zealand.
Genetics. 2018 May;209(1):65-76. doi: 10.1534/genetics.117.300627. Epub 2018 Feb 27.
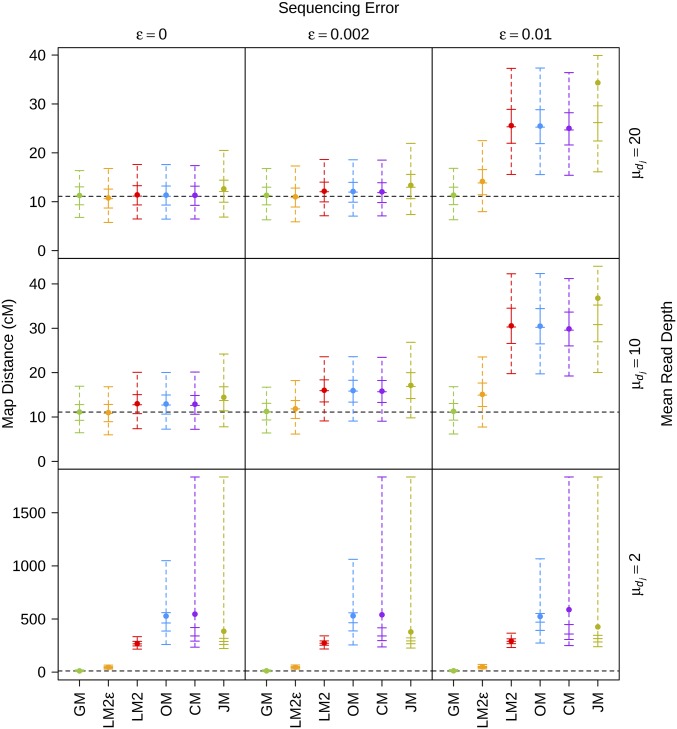
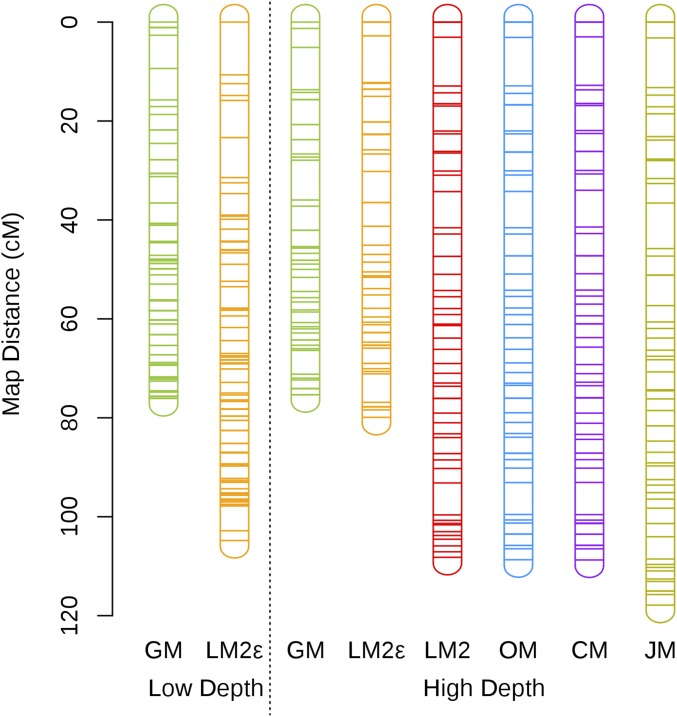
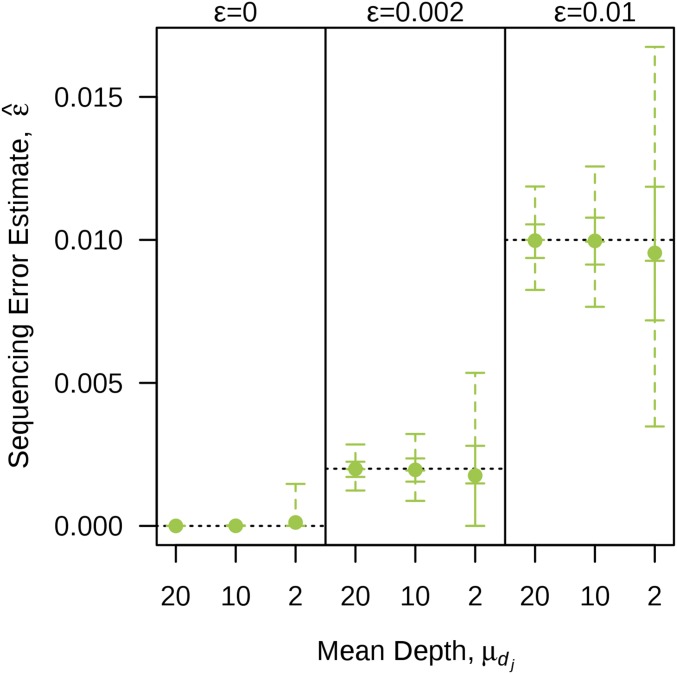
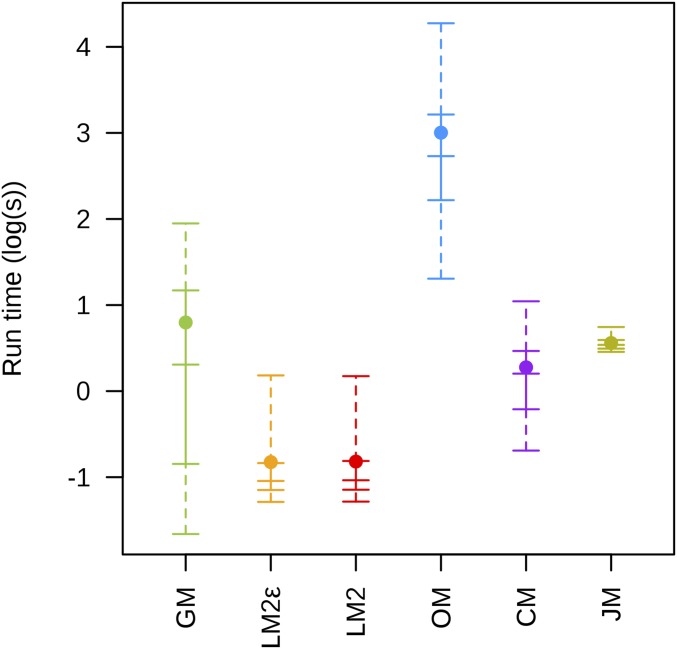
Next-generation sequencing is an efficient method that allows for substantially more markers than previous technologies, providing opportunities for building high-density genetic linkage maps, which facilitate the development of nonmodel species' genomic assemblies and the investigation of their genes. However, constructing genetic maps using data generated via high-throughput sequencing technology (, genotyping-by-sequencing) is complicated by the presence of sequencing errors and genotyping errors resulting from missing parental alleles due to low sequencing depth. If unaccounted for, these errors lead to inflated genetic maps. In addition, map construction in many species is performed using full-sibling family populations derived from the outcrossing of two individuals, where unknown parental phase and varying segregation types further complicate construction. We present a new methodology for modeling low coverage sequencing data in the construction of genetic linkage maps using full-sibling populations of diploid species, implemented in a package called GUSMap. Our model is based on the Lander-Green hidden Markov model but extended to account for errors present in sequencing data. We were able to obtain accurate estimates of the recombination fractions and overall map distance using GUSMap, while most existing mapping packages produced inflated genetic maps in the presence of errors. Our results demonstrate the feasibility of using low coverage sequencing data to produce genetic maps without requiring extensive filtering of potentially erroneous genotypes, provided that the associated errors are correctly accounted for in the model.
下一代测序是一种高效的方法,它可以比以前的技术产生更多的标记,为构建高密度遗传连锁图谱提供了机会,这有助于非模式物种基因组组装的发展和对其基因的研究。然而,使用高通量测序技术(如测序的基因分型)生成的数据构建遗传图谱会受到测序错误和由于测序深度低而导致的缺失亲本等位基因的基因分型错误的影响。如果不考虑这些错误,它们会导致遗传图谱膨胀。此外,许多物种的图谱构建都是使用来自两个个体杂交的全同胞家系群体进行的,其中未知的亲本相和不同的分离类型进一步增加了构建的复杂性。我们提出了一种新的方法,用于在使用二倍体物种的全同胞群体构建遗传连锁图谱时对低覆盖测序数据进行建模,该方法在一个名为 GUSMap 的软件包中实现。我们的模型基于 Lander-Green 隐马尔可夫模型,但扩展后可以考虑测序数据中的错误。我们能够使用 GUSMap 获得准确的重组分数和总体图谱距离估计,而大多数现有的映射软件包在存在错误的情况下会产生膨胀的遗传图谱。我们的结果表明,在模型中正确考虑相关错误的情况下,使用低覆盖测序数据生成遗传图谱而无需对潜在错误基因型进行广泛过滤是可行的。