Department of Microbiology and Immunology, The University of Melbourne at The Peter Doherty Institute for Infection and Immunity, Parkville, VIC 3010, Australia.
Cancer Biology And Therapy, Olivia Newton-John Cancer Research Institute, Heidelberg, VIC 3038, Australia.
Bioinformatics. 2023 Dec 1;39(12). doi: 10.1093/bioinformatics/btad685.
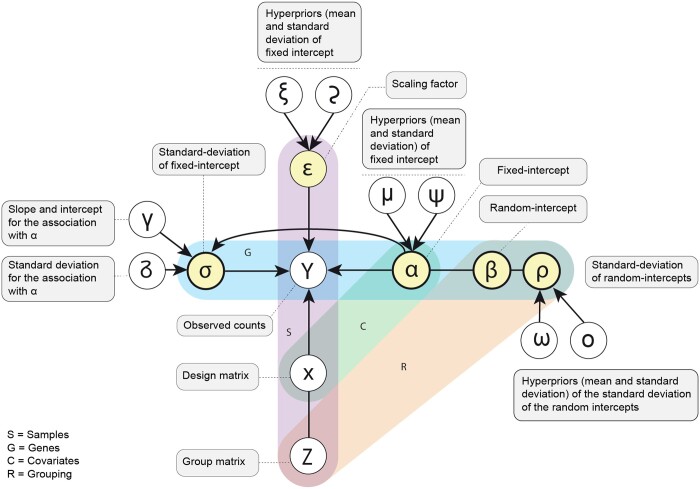
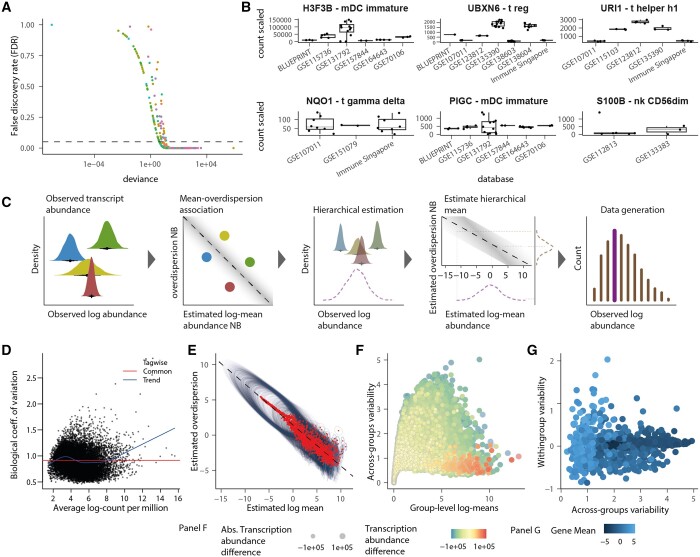
The precise characterization of cell-type transcriptomes is pivotal to understanding cellular lineages, deconvolution of bulk transcriptomes, and clinical applications. Single-cell RNA sequencing resources like the Human Cell Atlas have revolutionised cell-type profiling. However, challenges persist due to data heterogeneity and discrepancies across different studies. One limitation of prevailing tools such as CIBERSORTx is their inability to address hierarchical data structures and handle nonoverlapping gene sets across samples, relying on filtering or imputation.
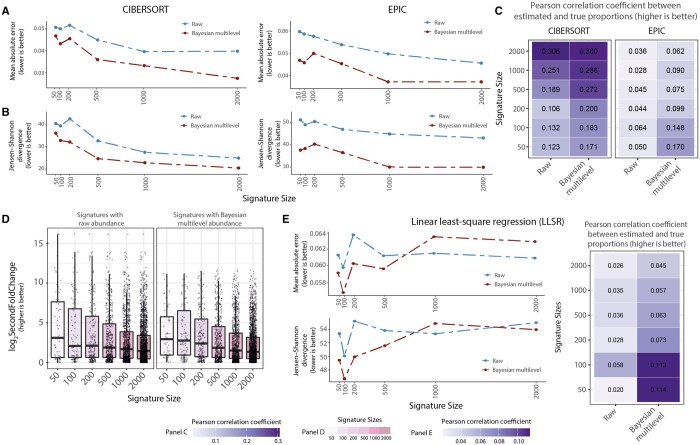
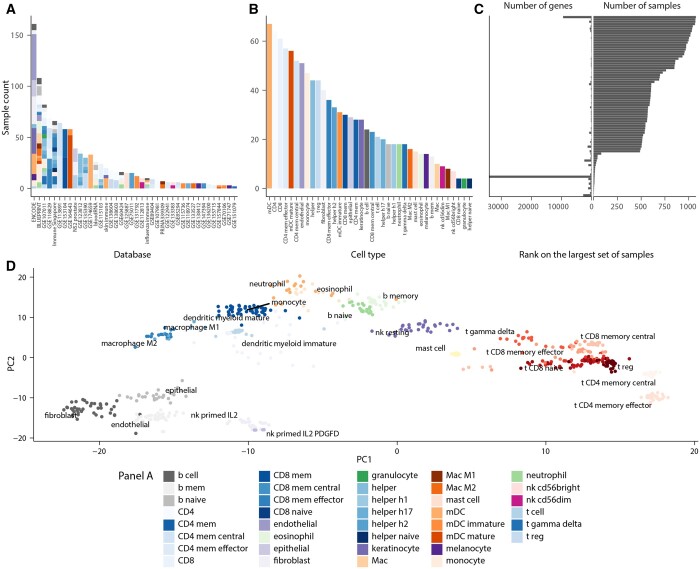
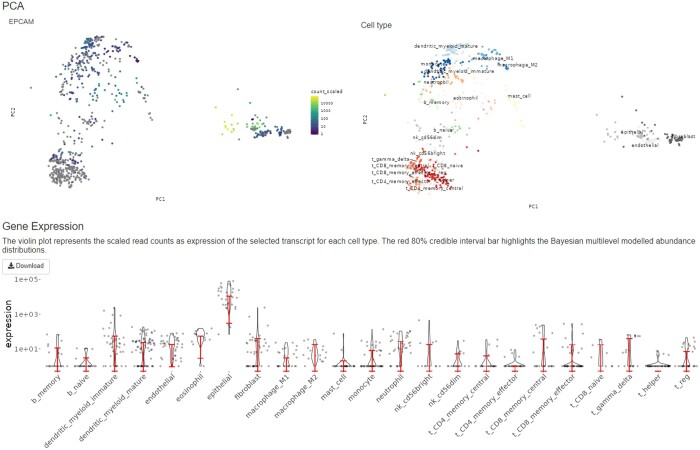
Here, we present cellsig, a Bayesian sparse multilevel model designed to improve signature estimation by adjusting data for multilevel effects and modelling for gene-set sparsity. Our model is tailored to large-scale, heterogeneous pseudobulk and bulk RNA sequencing data collections with nonoverlapping gene sets. We tested the performances of cellsig on a novel curated Human Bulk Cell-type Catalogue, which harmonizes 1435 samples across 58 datasets. We show that cellsig significantly enhances cell-type marker gene ranking performance. This approach is valuable for cell-type signature selection, with implications for marker gene validation, single-cell annotation, and deconvolution benchmarks.
Codes and the interactive app are available at https://github.com/stemangiola/cellsig; and the database is available at https://doi.org/10.5281/zenodo.7582421.
精确描述细胞类型的转录组对于理解细胞谱系、对大量转录组的反卷积以及临床应用至关重要。像人类细胞图谱这样的单细胞 RNA 测序资源已经彻底改变了细胞类型的分析。然而,由于数据的异质性以及不同研究之间的差异,仍然存在挑战。像 CIBERSORTx 这样的流行工具的一个限制是,它们无法解决层次数据结构的问题,并且无法处理样本之间非重叠的基因集,只能依靠过滤或插补。
在这里,我们提出了 cellsig,这是一种贝叶斯稀疏多层模型,旨在通过调整数据的多层次效应和基因集稀疏性来改善特征估计。我们的模型是针对具有非重叠基因集的大规模、异质的拟似和批量 RNA 测序数据集合量身定制的。我们在一个新的经过精心策划的人类批量细胞类型目录上测试了 cellsig 的性能,该目录协调了 58 个数据集的 1435 个样本。我们表明,cellsig 显著提高了细胞类型标记基因的排名性能。这种方法对于细胞类型特征选择很有价值,对标记基因验证、单细胞注释和反卷积基准测试具有重要意义。
代码和交互式应用程序可在 https://github.com/stemangiola/cellsig 上获得;数据库可在 https://doi.org/10.5281/zenodo.7582421 上获得。