Mangiola Stefano, Thomas Evan A, Modrák Martin, Vehtari Aki, Papenfuss Anthony T
The Walter and Eliza Hall Institute, Parkville, Victoria, 3052, Australia.
Institute of Microbiology of the Czech Academy of Sciences, Prague, 1083, Czech Republic.
NAR Genom Bioinform. 2021 Mar 1;3(1):lqab005. doi: 10.1093/nargab/lqab005. eCollection 2021 Mar.
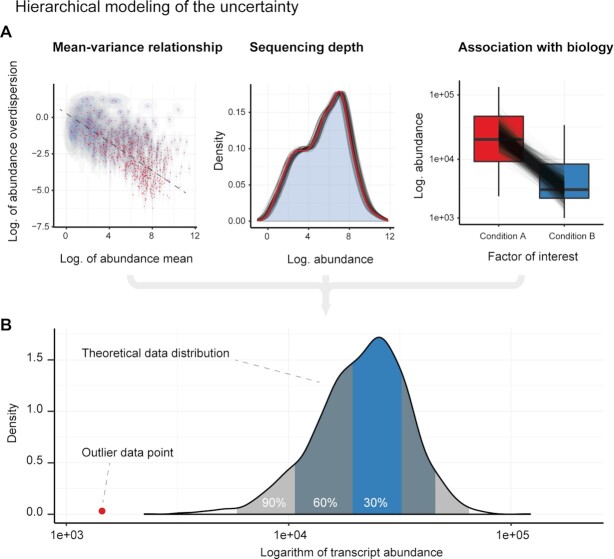
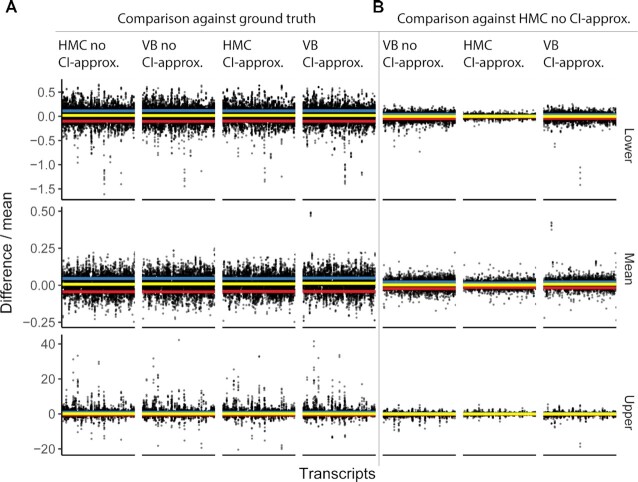
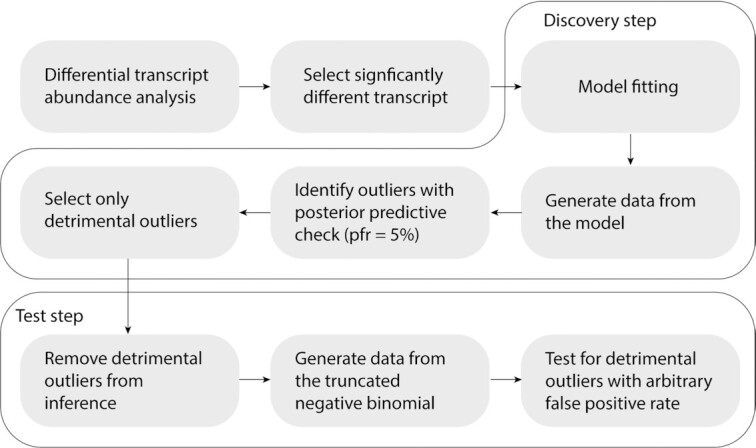
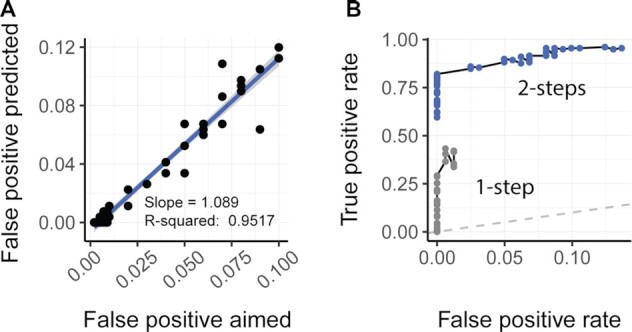
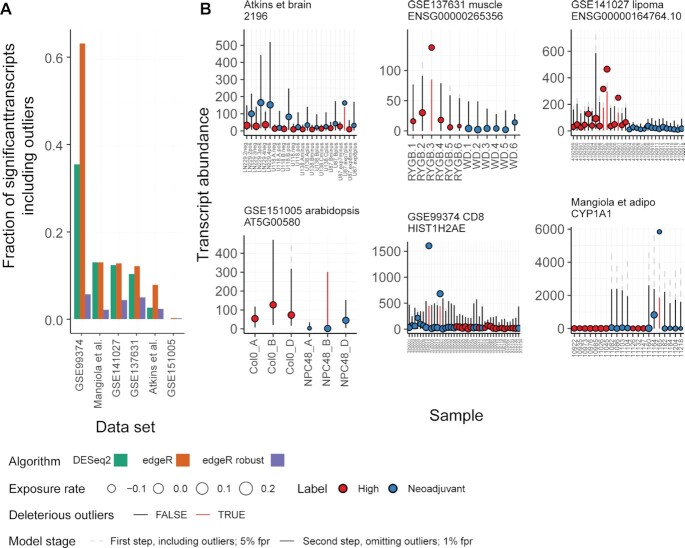
Relative transcript abundance has proven to be a valuable tool for understanding the function of genes in biological systems. For the differential analysis of transcript abundance using RNA sequencing data, the negative binomial model is by far the most frequently adopted. However, common methods that are based on a negative binomial model are not robust to extreme outliers, which we found to be abundant in public datasets. So far, no rigorous and probabilistic methods for detection of outliers have been developed for RNA sequencing data, leaving the identification mostly to visual inspection. Recent advances in Bayesian computation allow large-scale comparison of observed data against its theoretical distribution given in a statistical model. Here we propose ppcseq, a key quality-control tool for identifying transcripts that include outlier data points in differential expression analysis, which do not follow a negative binomial distribution. Applying ppcseq to analyse several publicly available datasets using popular tools, we show that from 3 to 10 percent of differentially abundant transcripts across algorithms and datasets had statistics inflated by the presence of outliers.
相对转录本丰度已被证明是了解生物系统中基因功能的一个有价值的工具。对于使用RNA测序数据进行转录本丰度的差异分析,负二项式模型是目前最常采用的。然而,基于负二项式模型的常用方法对极端异常值并不稳健,我们发现公共数据集中存在大量此类异常值。到目前为止,尚未开发出用于RNA测序数据的严格且概率性的异常值检测方法,异常值的识别大多依靠目视检查。贝叶斯计算的最新进展允许将观测数据与其在统计模型中给出的理论分布进行大规模比较。在此,我们提出了ppcseq,这是一种关键的质量控制工具,用于识别在差异表达分析中包含不符合负二项分布的异常数据点的转录本。使用流行工具将ppcseq应用于分析几个公开可用的数据集,我们发现,在不同算法和数据集中,有3%至10%的差异丰富转录本的统计量因异常值的存在而被夸大。