Hong Feng, Tian Lu, Devanarayan Viswanath
Takeda Pharmaceuticals, Cambridge, MA 02139, USA.
Department of Biomedical Data Science, Stanford University, Stanford, CA 94305, USA.
Mathematics (Basel). 2023 Feb;11(3). doi: 10.3390/math11030557. Epub 2023 Jan 20.
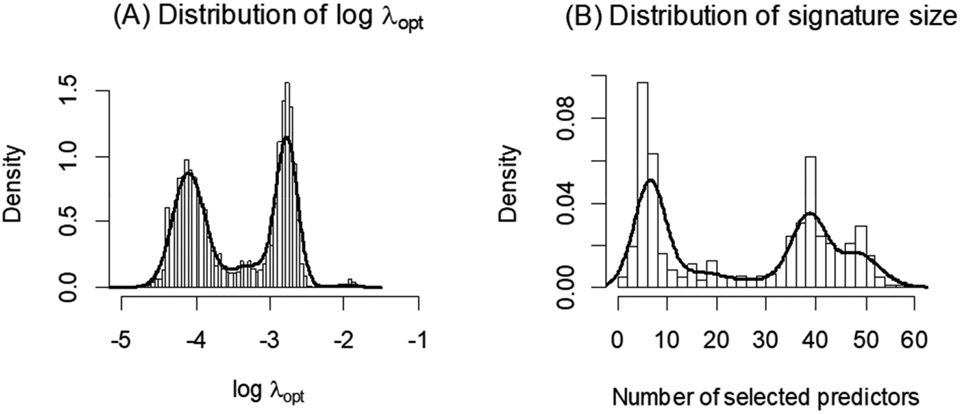
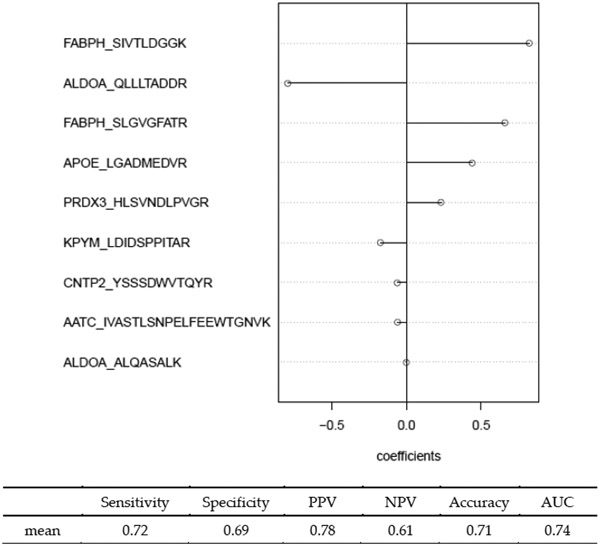
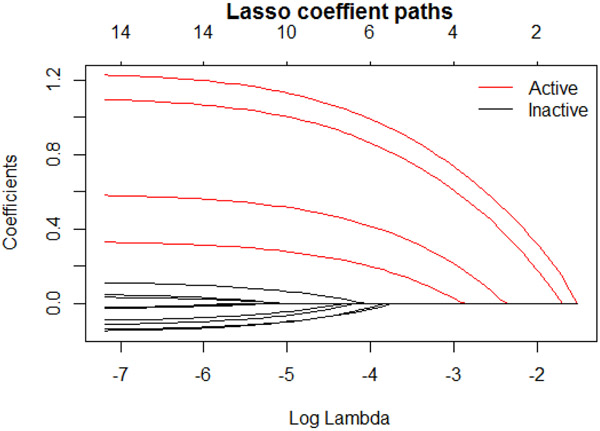
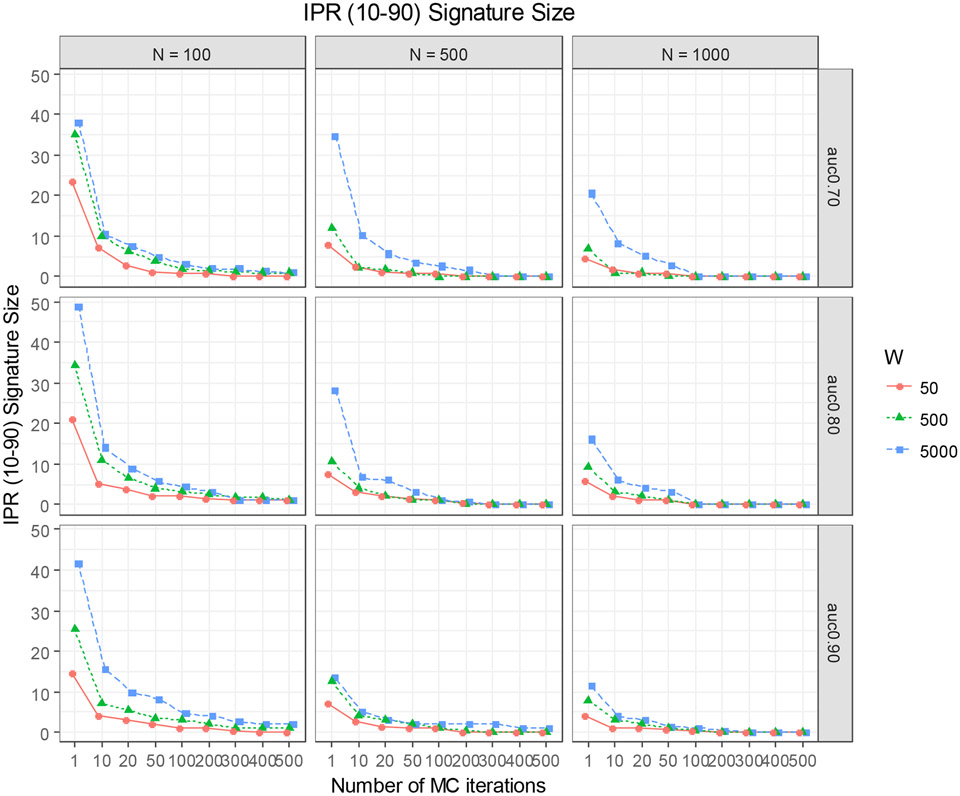
High-dimensional data applications often entail the use of various statistical and machine-learning algorithms to identify an optimal signature based on biomarkers and other patient characteristics that predicts the desired clinical outcome in biomedical research. Both the composition and predictive performance of such biomarker signatures are critical in various biomedical research applications. In the presence of a large number of features, however, a conventional regression analysis approach fails to yield a good prediction model. A widely used remedy is to introduce regularization in fitting the relevant regression model. In particular, a penalty on the regression coefficients is extremely useful, and very efficient numerical algorithms have been developed for fitting such models with different types of responses. This -based regularization tends to generate a parsimonious prediction model with promising prediction performance, i.e., feature selection is achieved along with construction of the prediction model. The variable selection, and hence the composition of the signature, as well as the prediction performance of the model depend on the choice of the penalty parameter used in the regularization. The penalty parameter is often chosen by K-fold cross-validation. However, such an algorithm tends to be unstable and may yield very different choices of the penalty parameter across multiple runs on the same dataset. In addition, the predictive performance estimates from the internal cross-validation procedure in this algorithm tend to be inflated. In this paper, we propose a Monte Carlo approach to improve the robustness of regularization parameter selection, along with an additional cross-validation wrapper for objectively evaluating the predictive performance of the final model. We demonstrate the improvements via simulations and illustrate the application via a real dataset.
高维数据应用通常需要使用各种统计和机器学习算法,以基于生物标志物和其他患者特征来识别最优特征集,从而在生物医学研究中预测期望的临床结果。此类生物标志物特征集的组成和预测性能在各种生物医学研究应用中都至关重要。然而,在存在大量特征的情况下,传统的回归分析方法无法产生良好的预测模型。一种广泛使用的补救方法是在拟合相关回归模型时引入正则化。特别是,对回归系数施加惩罚非常有用,并且已经开发出非常有效的数值算法来拟合具有不同类型响应的此类模型。这种基于惩罚的正则化倾向于生成具有良好预测性能的简约预测模型,即,在构建预测模型的同时实现了特征选择。变量选择以及因此特征集的组成,以及模型的预测性能取决于在惩罚正则化中使用的惩罚参数的选择。惩罚参数通常通过K折交叉验证来选择。然而,这样的算法往往不稳定,并且在同一数据集上多次运行时可能会产生非常不同的惩罚参数选择。此外,该算法中内部交叉验证过程的预测性能估计往往会被夸大。在本文中,我们提出了一种蒙特卡罗方法来提高正则化参数选择的稳健性,以及一个额外的交叉验证包装器,用于客观评估最终模型的预测性能。我们通过模拟展示了改进,并通过一个真实数据集说明了应用情况。