Nuffield Department of Medicine, Oxford University, Oxford, UK.
National Institute for Health Research (NIHR) Health Protection Research Unit on Healthcare Associated Infections and Antimicrobial Resistance at University of Oxford, Oxford, UK.
Microb Genom. 2023 Dec;9(12). doi: 10.1099/mgen.0.001151.
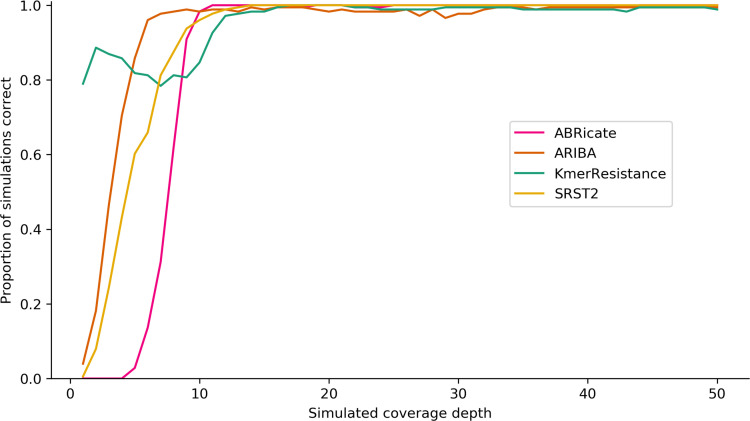
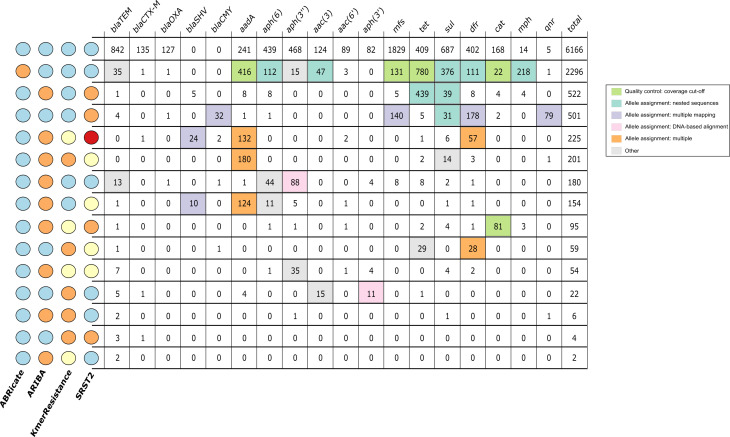
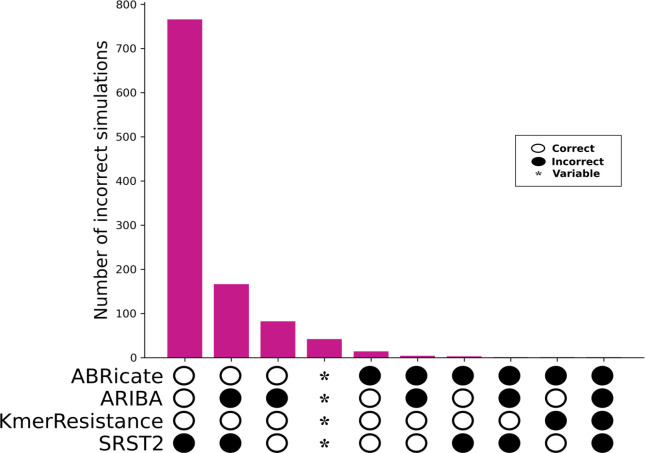
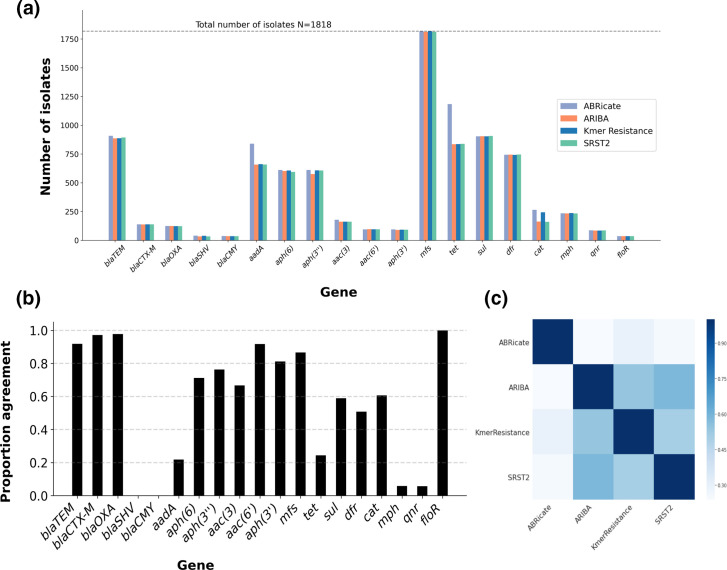
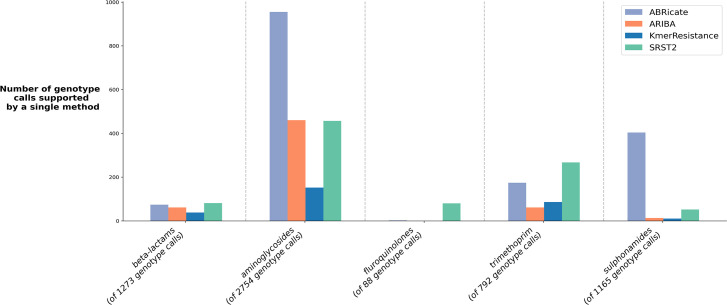
Several bioinformatics genotyping algorithms are now commonly used to characterize antimicrobial resistance (AMR) gene profiles in whole-genome sequencing (WGS) data, with a view to understanding AMR epidemiology and developing resistance prediction workflows using WGS in clinical settings. Accurately evaluating AMR in Enterobacterales, particularly , is of major importance, because this is a common pathogen. However, robust comparisons of different genotyping approaches on relevant simulated and large real-life WGS datasets are lacking. Here, we used both simulated datasets and a large set of real WGS data (=1818 isolates) to systematically investigate genotyping methods in greater detail. Simulated constructs and real sequences were processed using four different bioinformatic programs (ABRicate, ARIBA, KmerResistance and SRST2, run with the ResFinder database) and their outputs compared. For simulation tests where 3079 AMR gene variants were inserted into random sequence constructs, KmerResistance was correct for 3076 (99.9 %) simulations, ABRicate for 3054 (99.2 %), ARIBA for 2783 (90.4 %) and SRST2 for 2108 (68.5 %). For simulation tests where two closely related gene variants were inserted into random sequence constructs, KmerResistance identified the correct alleles in 35 338/46 318 (76.3 %) simulations, ABRicate identified them in 11 842/46 318 (25.6 %) simulations, ARIBA identified them in 1679/46 318 (3.6 %) simulations and SRST2 identified them in 2000/46 318 (4.3 %) simulations. In real data, across all methods, 1392/1818 (76 %) isolates had discrepant allele calls for at least 1 gene. In addition to highlighting areas for improvement in challenging scenarios, (e.g. identification of AMR genes at <10× coverage, identifying multiple closely related AMR genes present in the same sample), our evaluations identified some more systematic errors that could be readily soluble, such as repeated misclassification (i.e. naming) of genes as shorter variants of the same gene present within the reference resistance gene database. Such naming errors accounted for at least 2530/4321 (59 %) of the discrepancies seen in real data. Moreover, many of the remaining discrepancies were likely 'artefactual', with reporting of cut-off differences accounting for at least 1430/4321 (33 %) discrepants. Whilst we found that comparing outputs generated by running multiple algorithms on the same dataset could identify and resolve these algorithmic artefacts, the results of our evaluations emphasize the need for developing new and more robust genotyping algorithms to further improve accuracy and performance.
目前,有几种生物信息学基因分型算法常用于全基因组测序 (WGS) 数据中抗菌药物耐药 (AMR) 基因谱的特征描述,旨在了解 AMR 流行病学,并在临床环境中使用 WGS 开发耐药性预测工作流程。正确评估肠杆菌科中的 AMR 尤其重要,因为这是一种常见的病原体。然而,不同基因分型方法在相关模拟和大型真实 WGS 数据集上的稳健比较仍然缺乏。在这里,我们使用模拟数据集和一大组真实的 WGS 数据(= 1818 株)更详细地系统研究基因分型方法。使用四个不同的生物信息学程序(ABRicate、ARIBA、KmerResistance 和 SRST2,使用 ResFinder 数据库运行)处理模拟构建体和真实序列,并比较它们的输出。对于将 3079 个 AMR 基因变体插入随机序列构建体的模拟测试,KmerResistance 正确 3076(99.9%)次模拟,ABRicate 正确 3054(99.2%)次模拟,ARIBA 正确 2783(90.4%)次模拟,SRST2 正确 2108(68.5%)次模拟。对于将两个密切相关的基因变体插入随机序列构建体的模拟测试,KmerResistance 在 35338/46318(76.3%)次模拟中正确识别了正确的等位基因,ABRicate 在 11842/46318(25.6%)次模拟中正确识别了正确的等位基因,ARIBA 在 1679/46318(3.6%)次模拟中正确识别了正确的等位基因,而 SRST2 在 2000/46318(4.3%)次模拟中正确识别了正确的等位基因。在真实数据中,所有方法中,1392/1818(76%)株至少有 1 个基因的等位基因检测结果不一致。除了突出在具有挑战性的情况下需要改进的领域(例如,在<10×覆盖的情况下识别 AMR 基因,识别同一样本中存在的多个密切相关的 AMR 基因)外,我们的评估还确定了一些更系统的错误,这些错误很容易解决,例如,将基因重复错误分类(即命名)为同一参考耐药基因数据库中存在的相同基因的较短变体。这种命名错误占真实数据中观察到的差异的至少 2530/4321(59%)。此外,许多剩余的差异可能是“人为的”,报告截止值差异至少占 1430/4321(33%)的差异。虽然我们发现通过在同一数据集上运行多个算法来比较输出可以识别和解决这些算法错误,但我们的评估结果强调需要开发新的、更强大的基因分型算法,以进一步提高准确性和性能。