Department of Pathology, University of Pittsburgh, Pittsburgh, United States.
High Throughput Genome Center, University of Pittsburgh, Pittsburgh, United States.
Elife. 2024 Jan 11;12:RP87607. doi: 10.7554/eLife.87607.
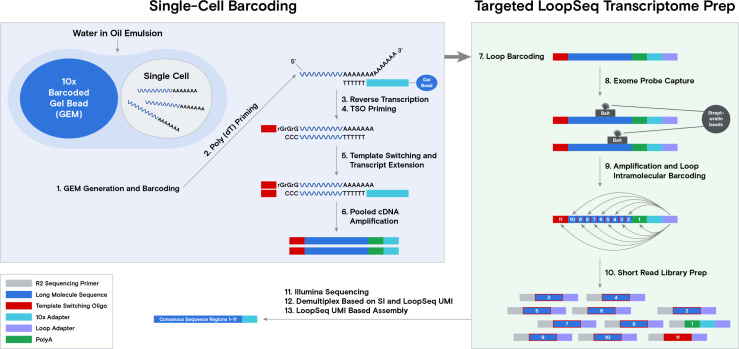
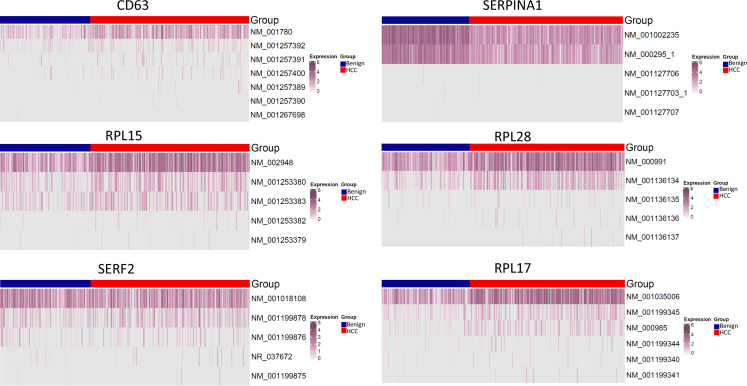
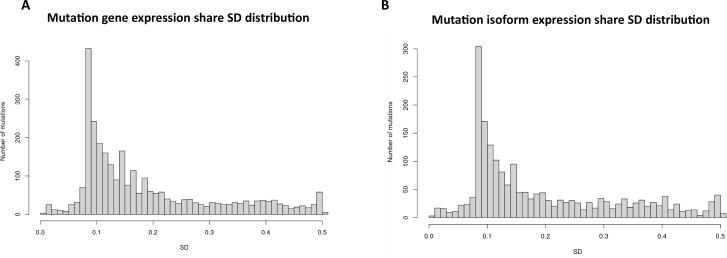
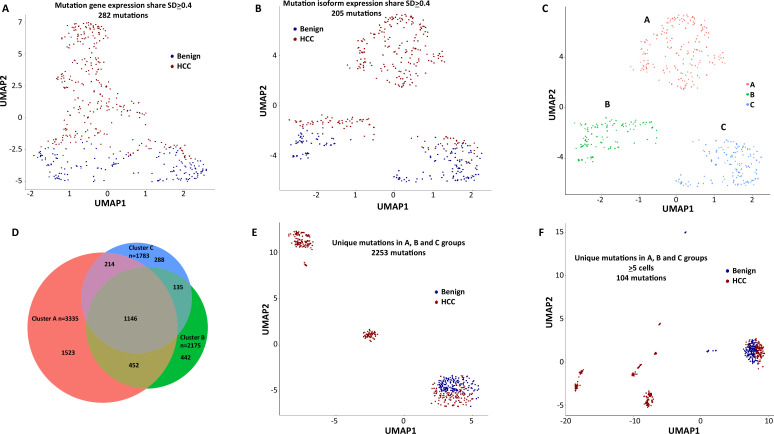
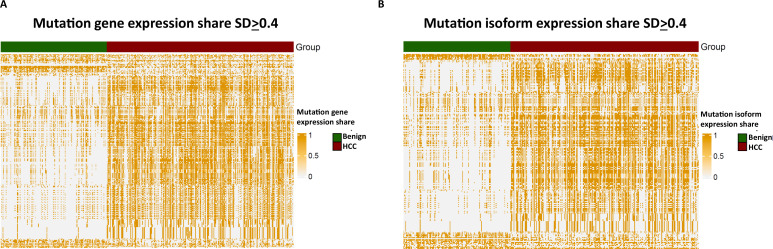
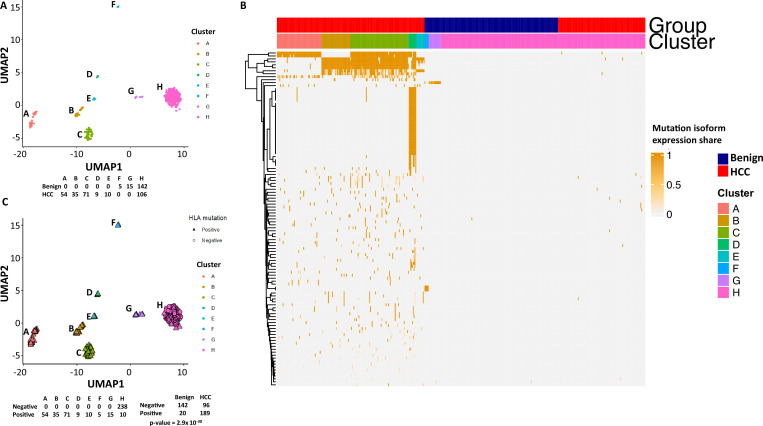
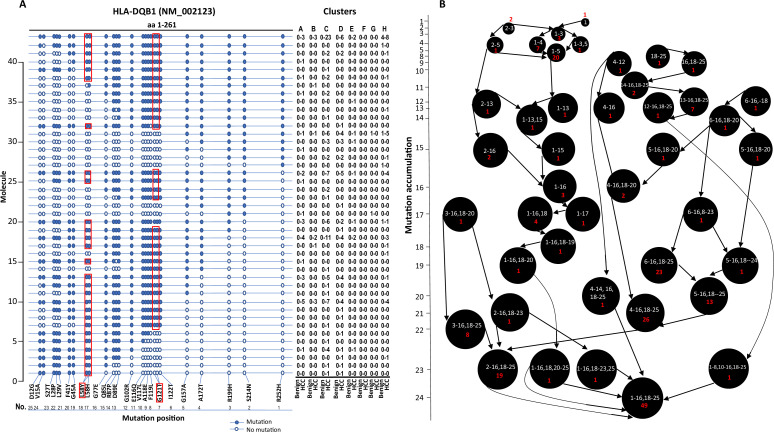
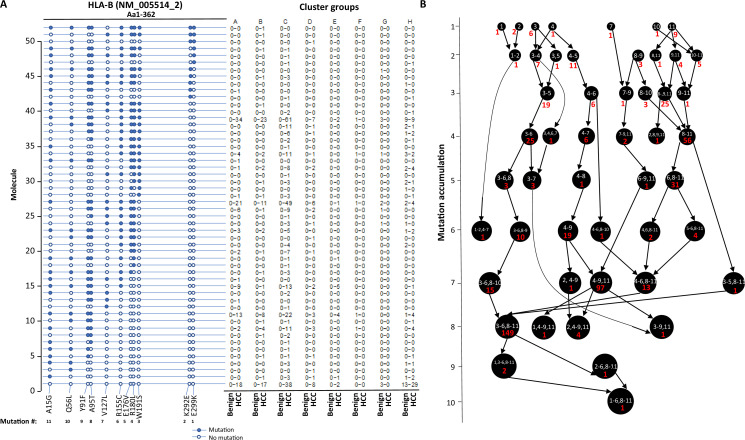
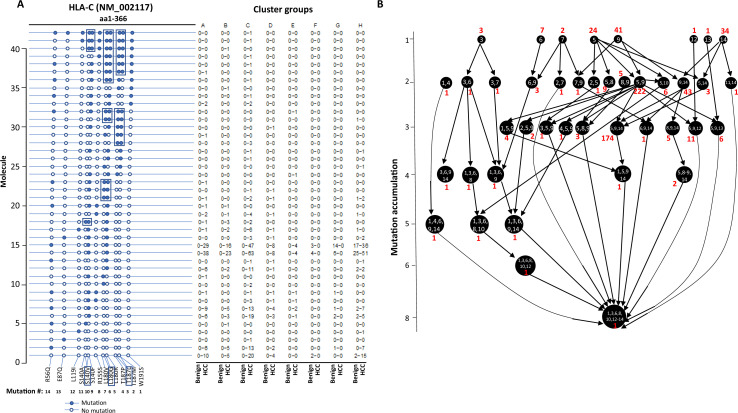
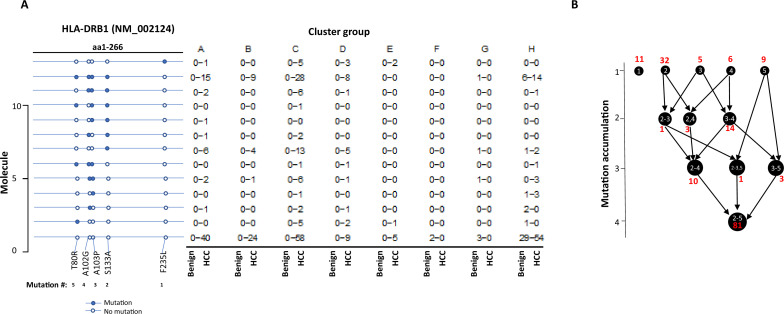
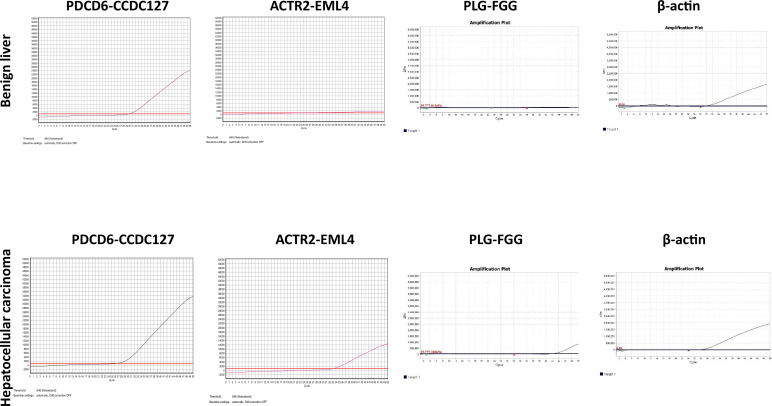
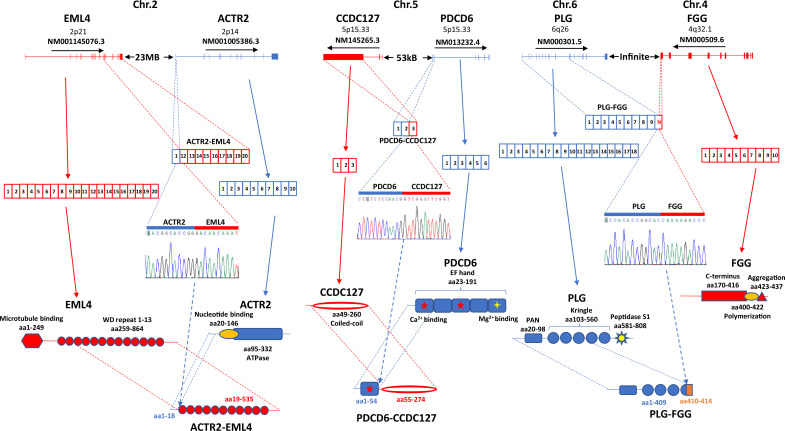
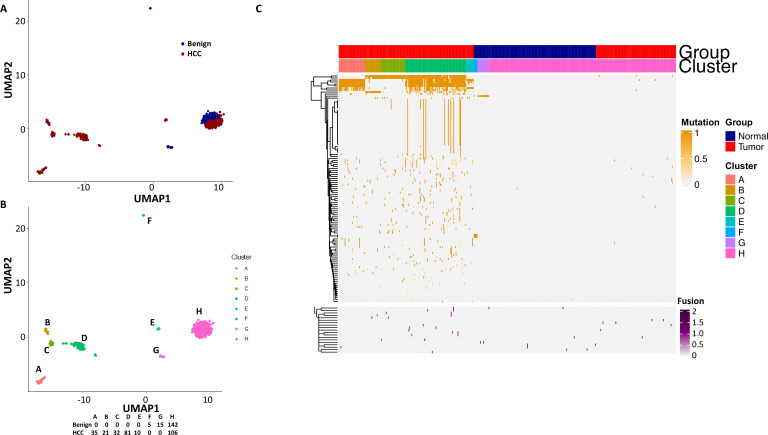
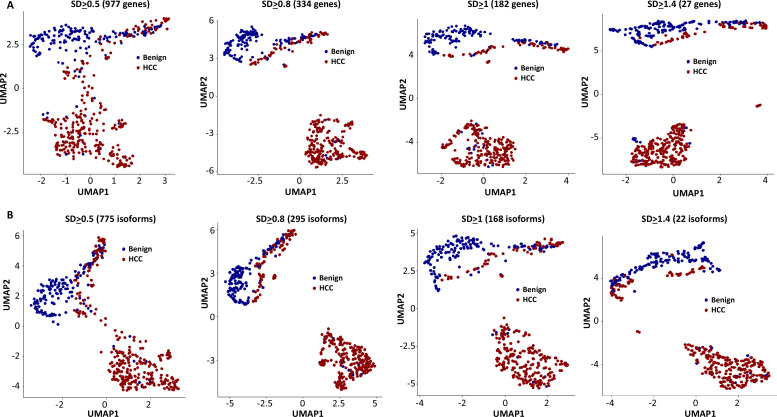
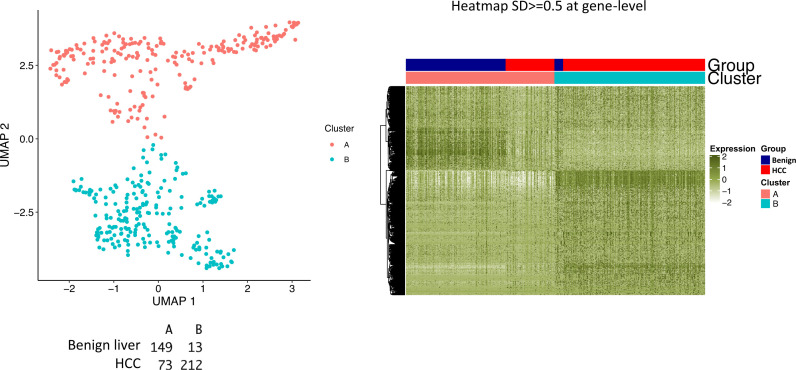
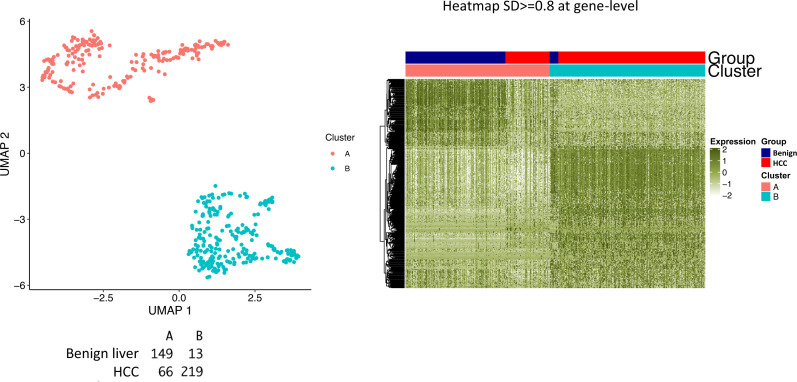
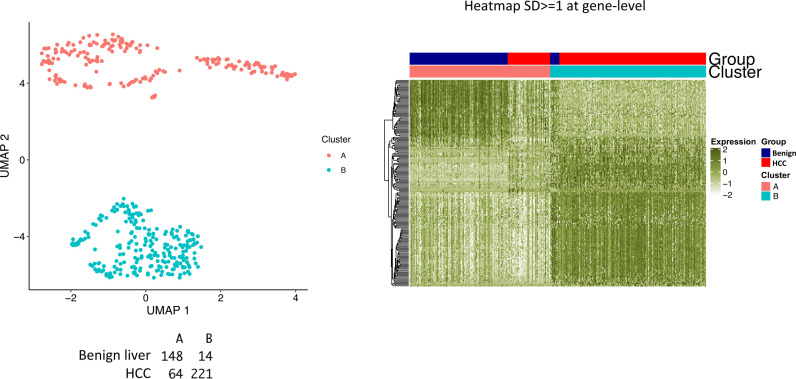
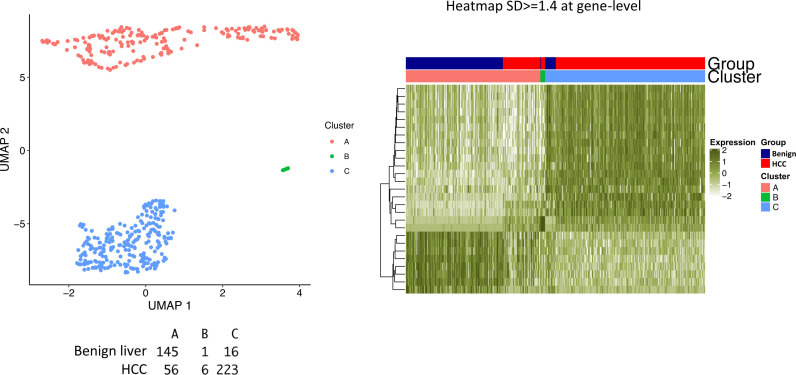
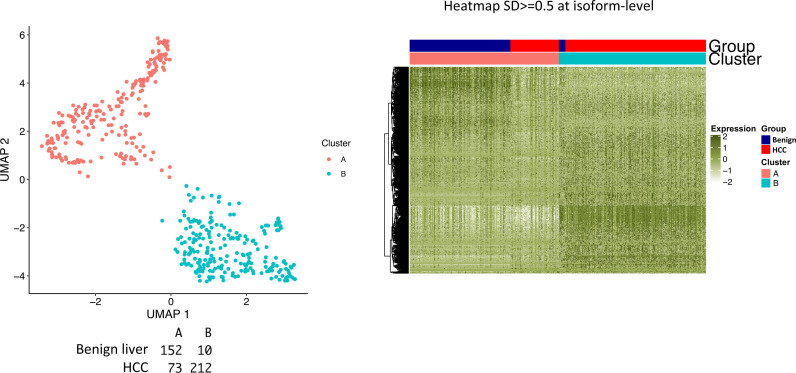
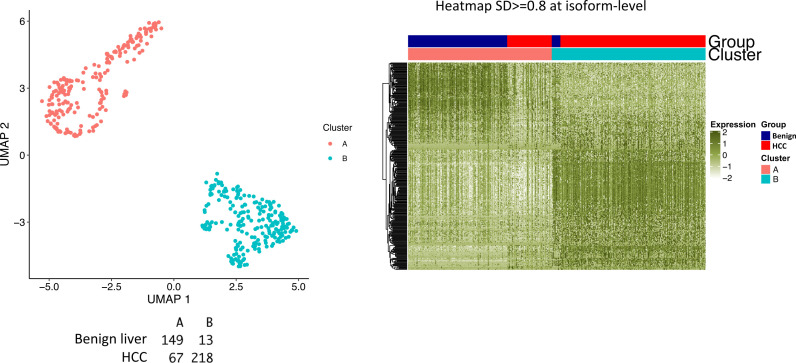
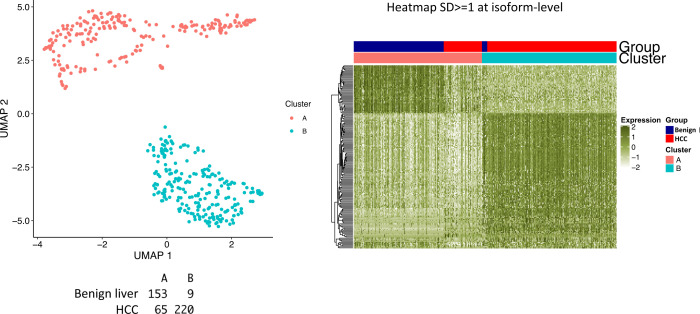
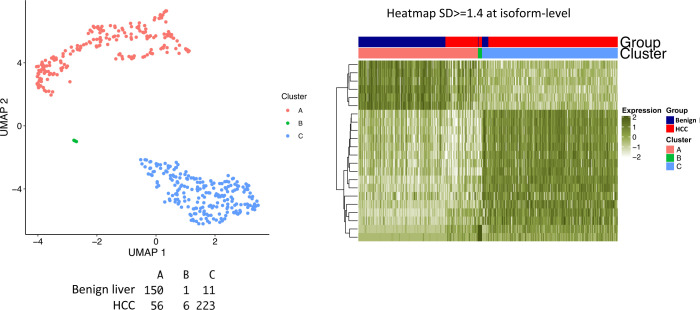
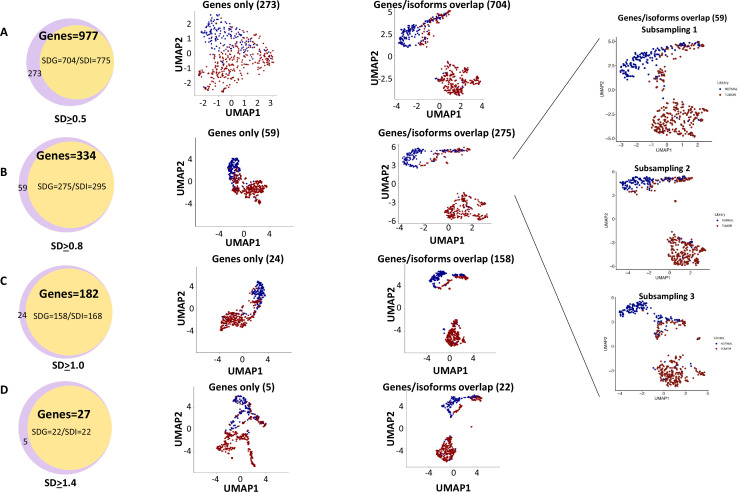
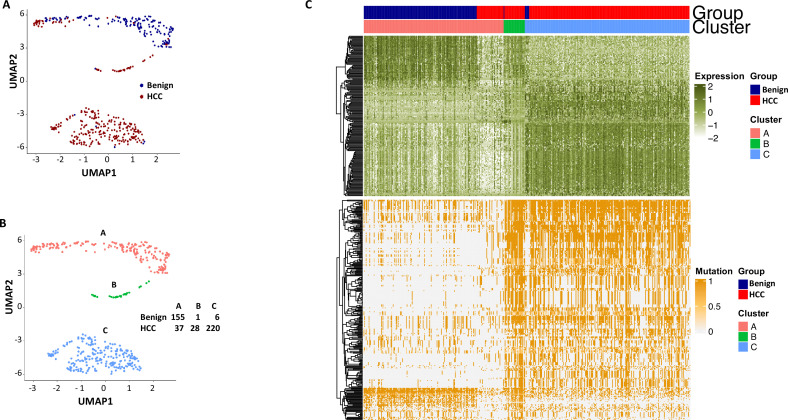
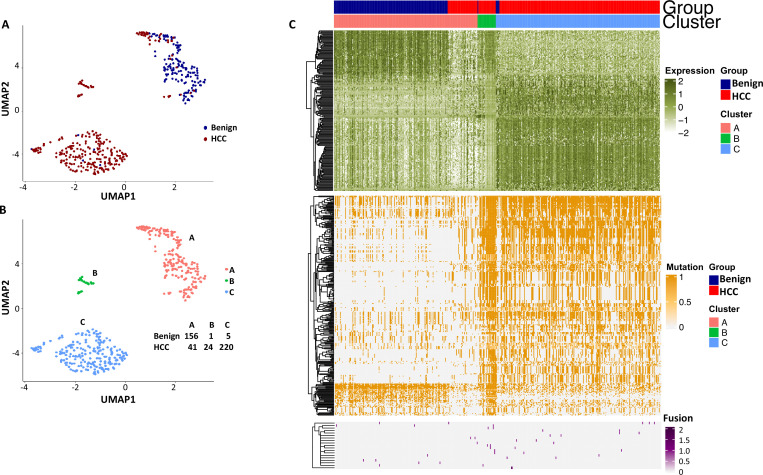
The protein diversity of mammalian cells is determined by arrays of isoforms from genes. Genetic mutation is essential in species evolution and cancer development. Accurate long-read transcriptome sequencing at single-cell level is required to decipher the spectrum of protein expressions in mammalian organisms. In this report, we developed a synthetic long-read single-cell sequencing technology based on LOOPSeq technique. We applied this technology to analyze 447 transcriptomes of hepatocellular carcinoma (HCC) and benign liver from an individual. Through Uniform Manifold Approximation and Projection analysis, we identified a panel of mutation mRNA isoforms highly specific to HCC cells. The evolution pathways that led to the hyper-mutation clusters in single human leukocyte antigen molecules were identified. Novel fusion transcripts were detected. The combination of gene expressions, fusion gene transcripts, and mutation gene expressions significantly improved the classification of liver cancer cells versus benign hepatocytes. In conclusion, LOOPSeq single-cell technology may hold promise to provide a new level of precision analysis on the mammalian transcriptome.
哺乳动物细胞的蛋白质多样性是由基因的同工型阵列决定的。遗传突变是物种进化和癌症发展的关键。为了解哺乳动物生物体内蛋白质表达的全貌,需要在单细胞水平上进行准确的长读长转录组测序。在本报告中,我们开发了一种基于 LOOPSeq 技术的合成长读长单细胞测序技术。我们将该技术应用于分析来自个体的 447 个肝癌 (HCC) 和良性肝的转录组。通过一致流形逼近和投影分析,我们鉴定了一组高度特异于 HCC 细胞的突变 mRNA 同工型。鉴定了导致单个人类白细胞抗原分子中超突变簇的进化途径。检测到新的融合转录本。基因表达、融合基因转录本和突变基因表达的组合显著提高了肝癌细胞与良性肝细胞的分类。总之,LOOPSeq 单细胞技术有望为哺乳动物转录组提供新的精度分析水平。