MRC Laboratory of Molecular Biology, Cambridge, UK.
Science for Life Laboratory, KTH Royal Institute of Technology, Stockholm, Sweden.
Nature. 2024 Apr;628(8007):450-457. doi: 10.1038/s41586-024-07215-4. Epub 2024 Feb 26.
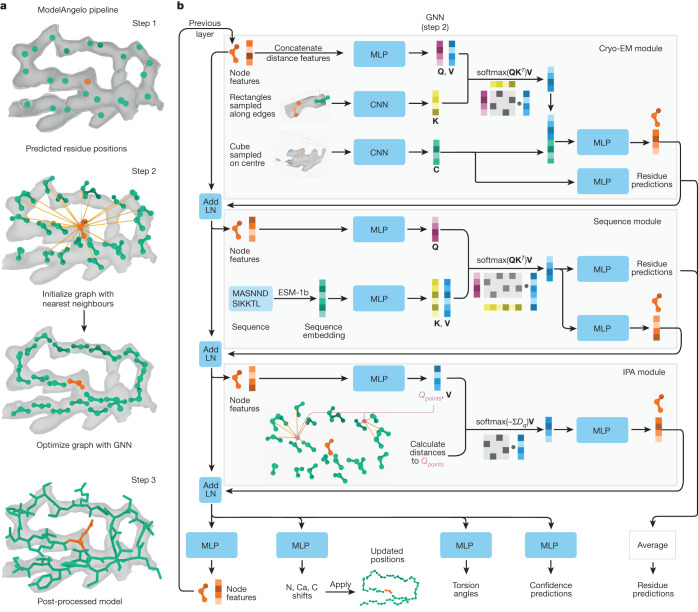
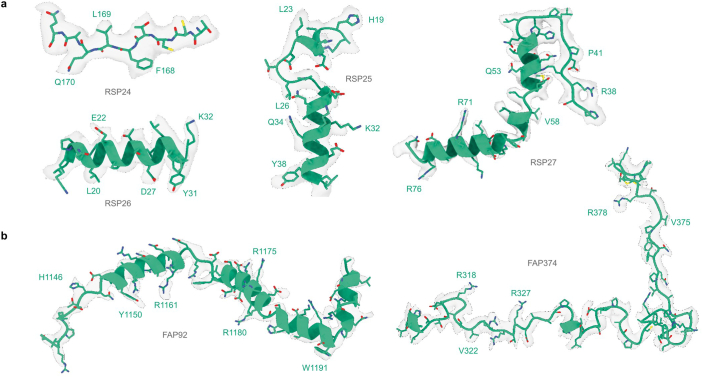
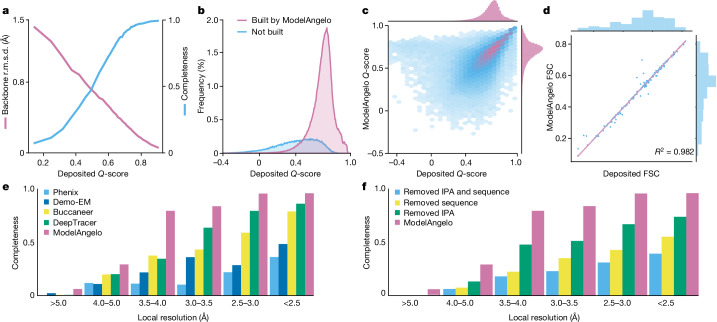
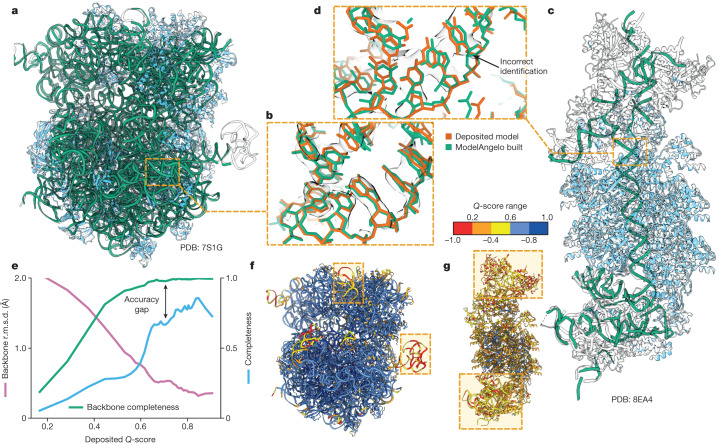
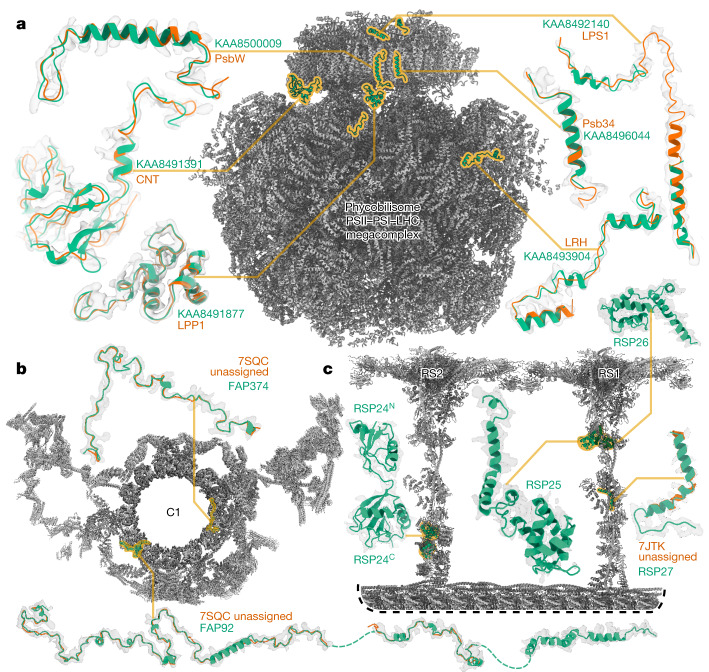
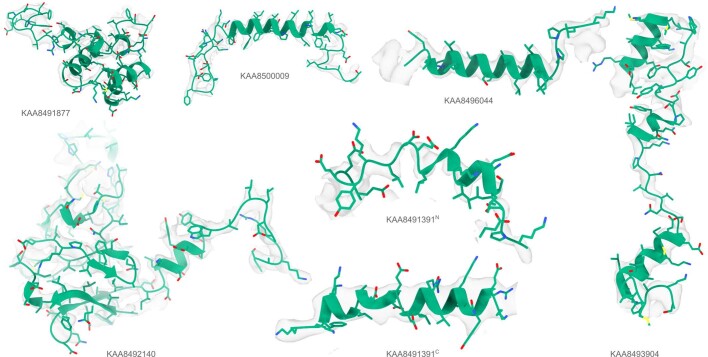
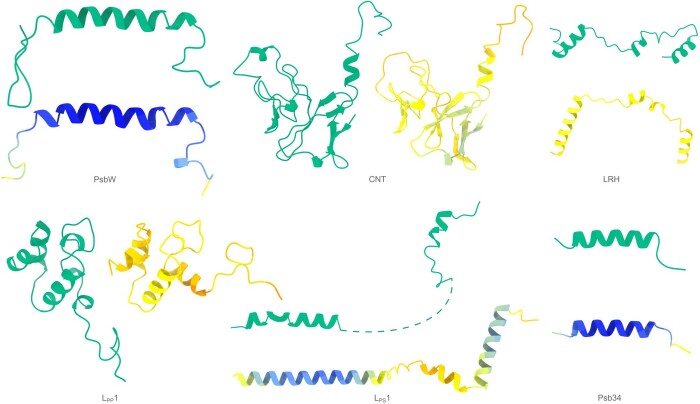
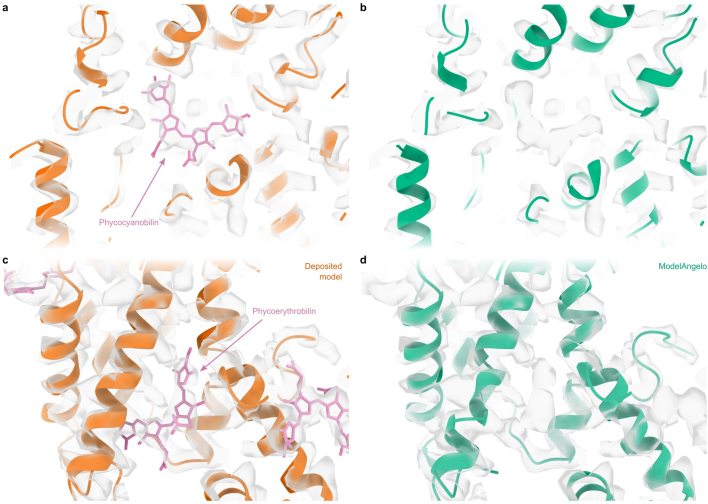
Interpreting electron cryo-microscopy (cryo-EM) maps with atomic models requires high levels of expertise and labour-intensive manual intervention in three-dimensional computer graphics programs. Here we present ModelAngelo, a machine-learning approach for automated atomic model building in cryo-EM maps. By combining information from the cryo-EM map with information from protein sequence and structure in a single graph neural network, ModelAngelo builds atomic models for proteins that are of similar quality to those generated by human experts. For nucleotides, ModelAngelo builds backbones with similar accuracy to those built by humans. By using its predicted amino acid probabilities for each residue in hidden Markov model sequence searches, ModelAngelo outperforms human experts in the identification of proteins with unknown sequences. ModelAngelo will therefore remove bottlenecks and increase objectivity in cryo-EM structure determination.
用原子模型解释电子晶体学显微镜 (cryo-EM) 图谱需要高水平的专业知识和三维计算机图形程序中的劳动密集型手动干预。在这里,我们展示了 ModelAngelo,这是一种用于 cryo-EM 图谱中自动构建原子模型的机器学习方法。通过将 cryo-EM 图谱中的信息与蛋白质序列和结构中的信息结合在单个图神经网络中,ModelAngelo 为具有相似质量的蛋白质构建原子模型,这些模型与人类专家生成的模型相当。对于核苷酸,ModelAngelo 构建的骨架与人类构建的骨架具有相似的准确性。通过在隐马尔可夫模型序列搜索中使用其预测的每个残基的氨基酸概率,ModelAngelo 在识别具有未知序列的蛋白质方面优于人类专家。因此,ModelAngelo 将消除 cryo-EM 结构测定中的瓶颈并提高客观性。