Department of Gastroenterology, Changhai Hospital, Naval Medical University, 168 Changhai Road, Shanghai, 200433, China.
Shanghai Institute of Pancreatic Diseases, Shanghai, China.
Hum Genomics. 2024 Feb 27;18(1):21. doi: 10.1186/s40246-024-00586-9.
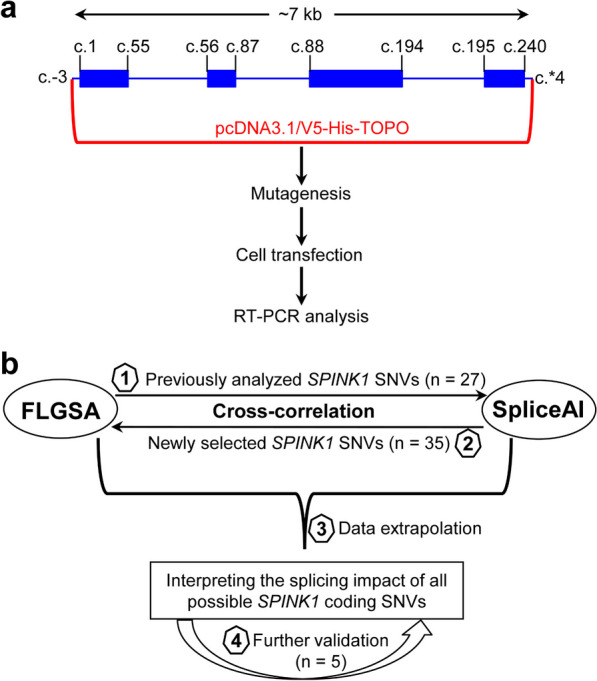
Single-nucleotide variants (SNVs) within gene coding sequences can significantly impact pre-mRNA splicing, bearing profound implications for pathogenic mechanisms and precision medicine. In this study, we aim to harness the well-established full-length gene splicing assay (FLGSA) in conjunction with SpliceAI to prospectively interpret the splicing effects of all potential coding SNVs within the four-exon SPINK1 gene, a gene associated with chronic pancreatitis.
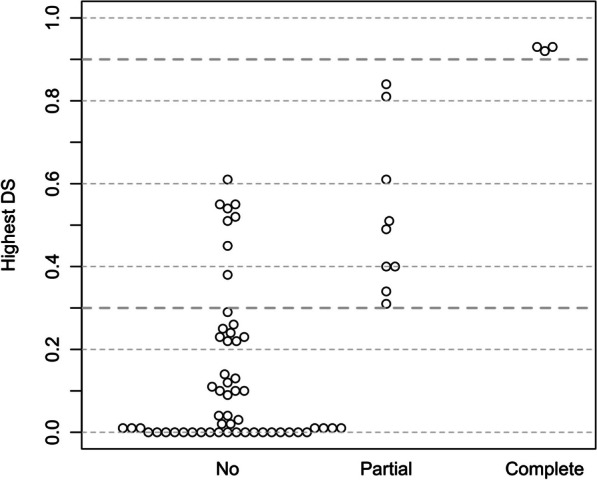
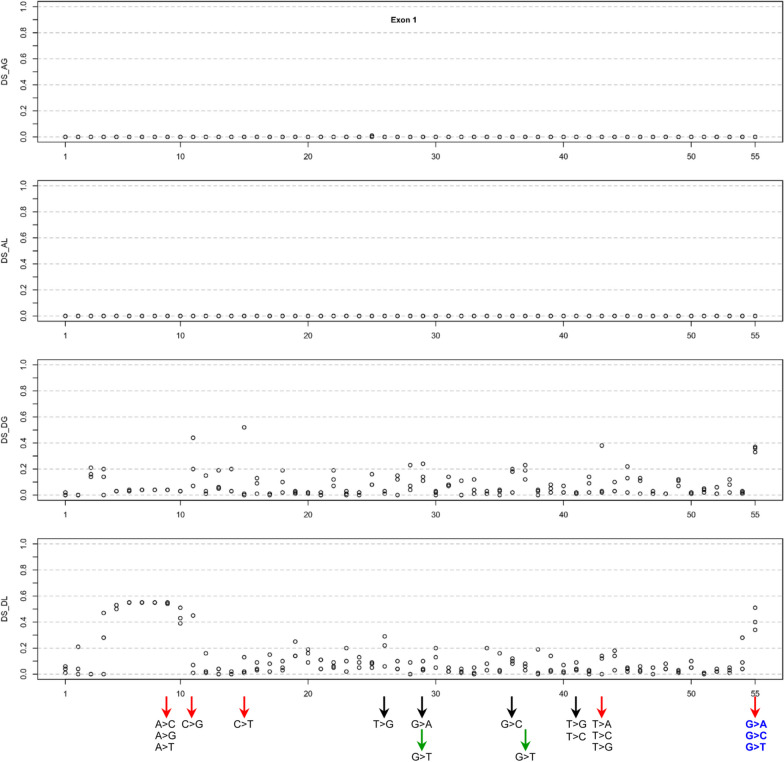
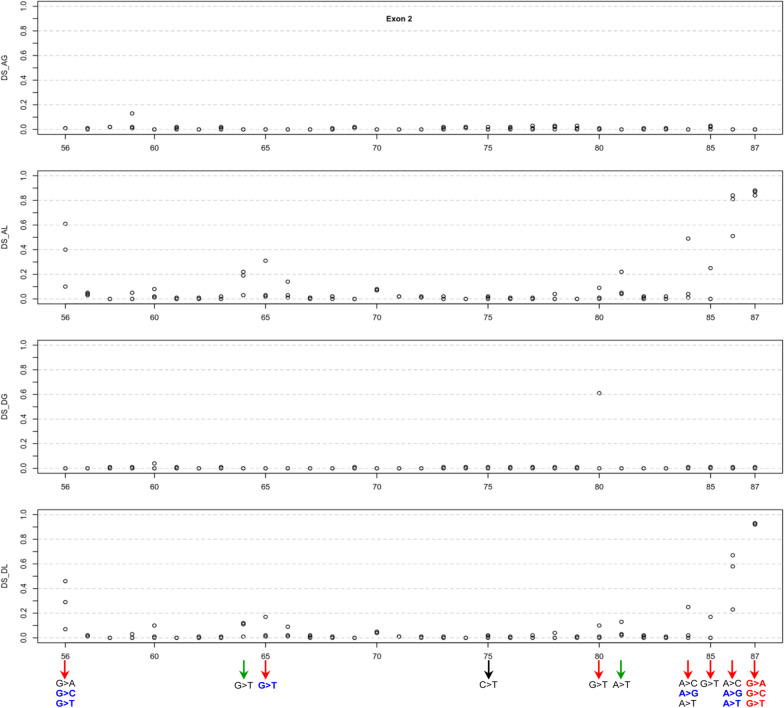
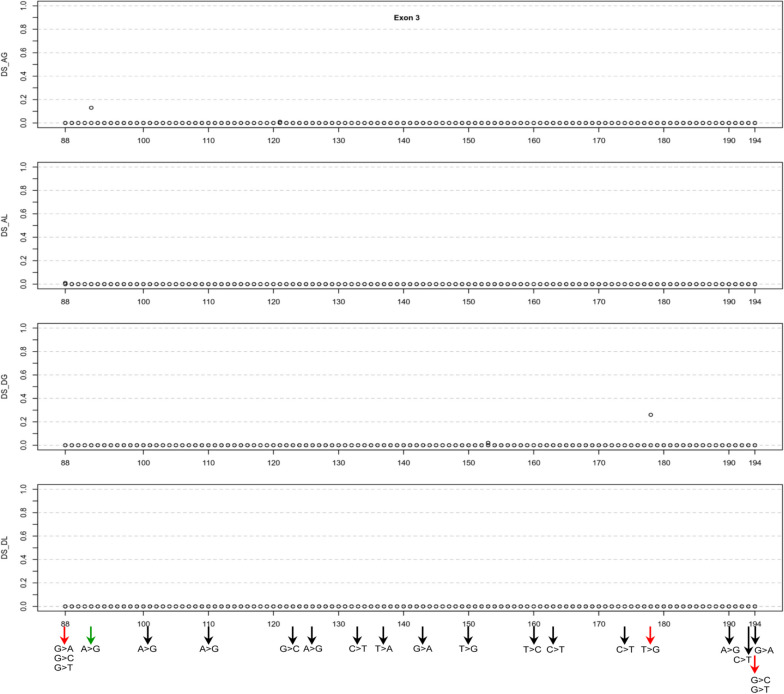
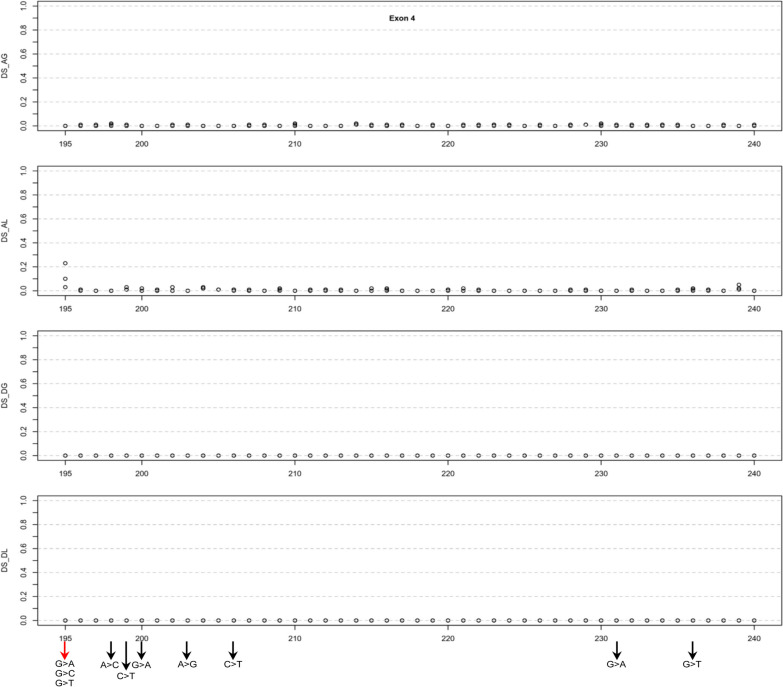
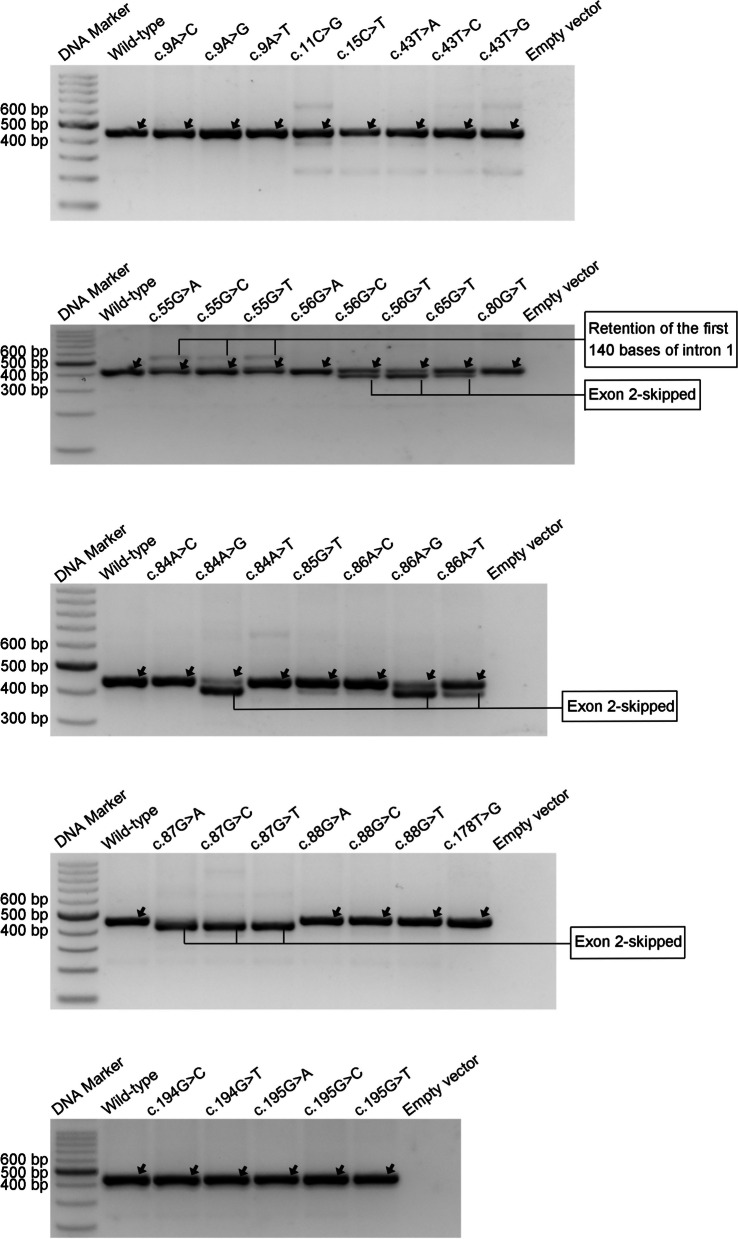
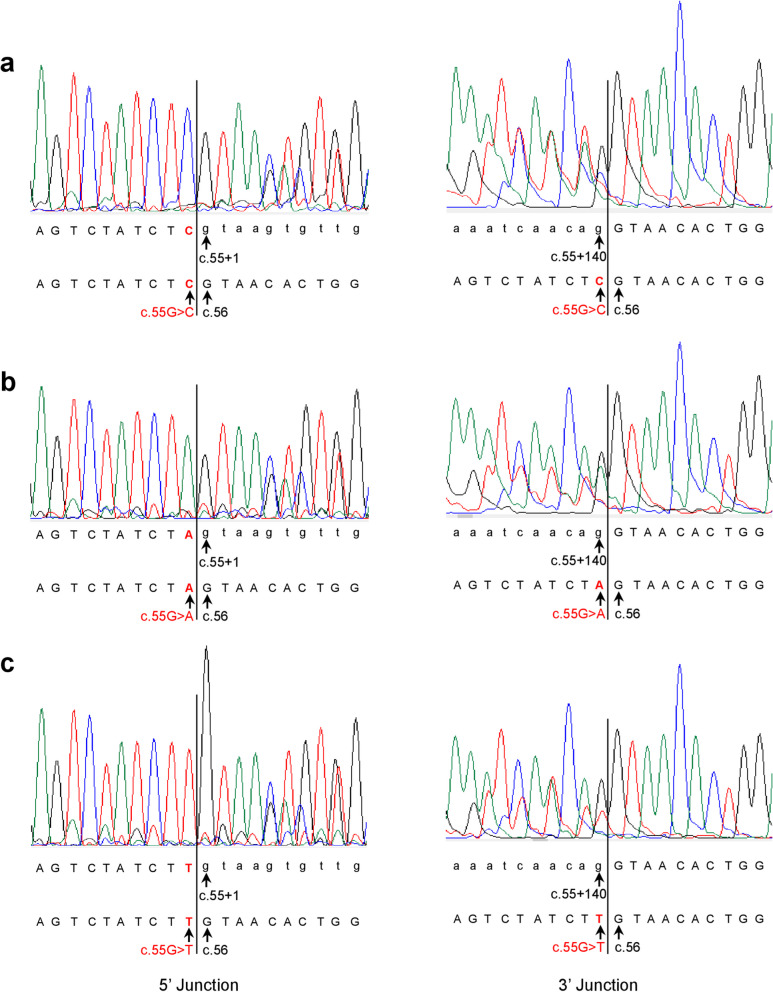
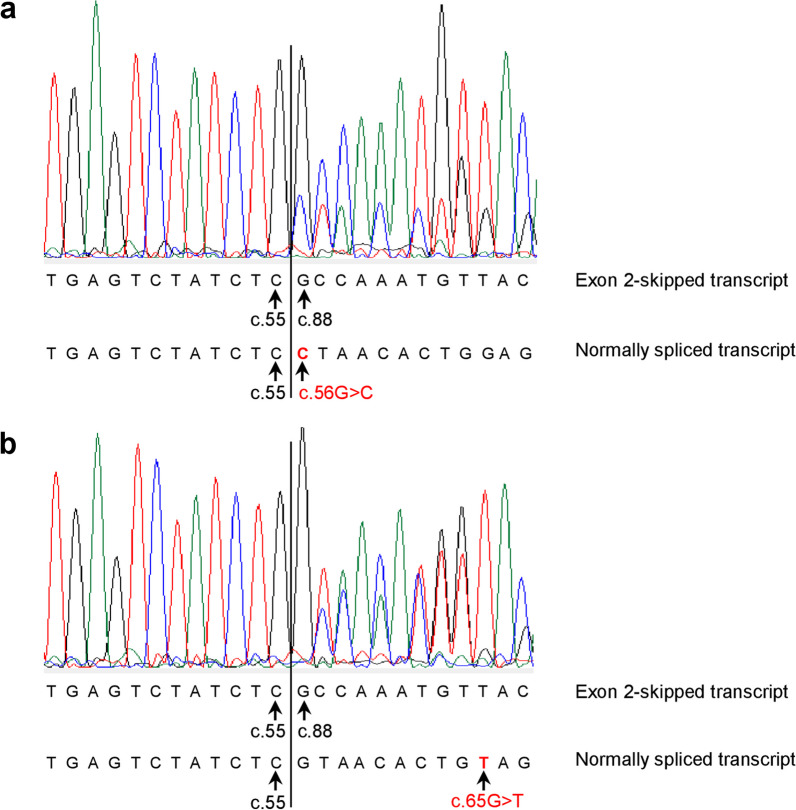
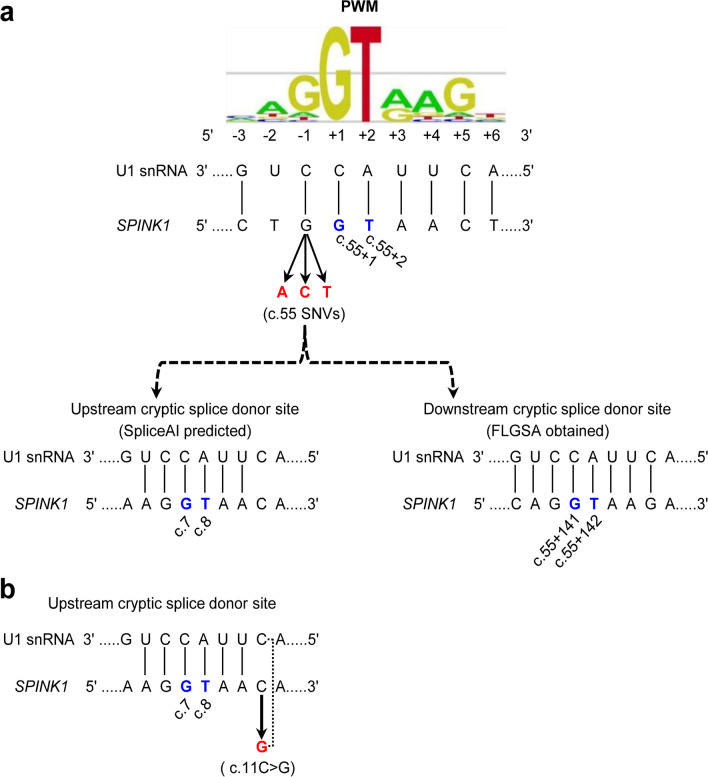
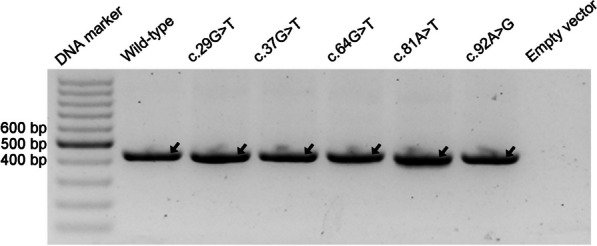
Our study began with a retrospective analysis of 27 SPINK1 coding SNVs previously assessed using FLGSA, proceeded with a prospective analysis of 35 new FLGSA-tested SPINK1 coding SNVs, followed by data extrapolation, and ended with further validation. In total, we analyzed 67 SPINK1 coding SNVs, which account for 9.3% of the 720 possible coding SNVs. Among these 67 FLGSA-analyzed SNVs, 12 were found to impact splicing. Through detailed comparison of FLGSA results and SpliceAI predictions, we inferred that the remaining 653 untested coding SNVs in the SPINK1 gene are unlikely to significantly affect splicing. Of the 12 splice-altering events, nine produced both normally spliced and aberrantly spliced transcripts, while the remaining three only generated aberrantly spliced transcripts. These splice-impacting SNVs were found solely in exons 1 and 2, notably at the first and/or last coding nucleotides of these exons. Among the 12 splice-altering events, 11 were missense variants (2.17% of 506 potential missense variants), and one was synonymous (0.61% of 164 potential synonymous variants). Notably, adjusting the SpliceAI cut-off to 0.30 instead of the conventional 0.20 would improve specificity without reducing sensitivity.
By integrating FLGSA with SpliceAI, we have determined that less than 2% (1.67%) of all possible coding SNVs in SPINK1 significantly influence splicing outcomes. Our findings emphasize the critical importance of conducting splicing analysis within the broader genomic sequence context of the study gene and highlight the inherent uncertainties associated with intermediate SpliceAI scores (0.20 to 0.80). This study contributes to the field by being the first to prospectively interpret all potential coding SNVs in a disease-associated gene with a high degree of accuracy, representing a meaningful attempt at shifting from retrospective to prospective variant analysis in the era of exome and genome sequencing.
基因编码序列中的单核苷酸变异(SNVs)可显著影响前体 mRNA 的剪接,对致病机制和精准医学具有深远意义。本研究旨在结合全长基因剪接分析(FLGSA)和 SpliceAI,前瞻性地解释与慢性胰腺炎相关的 SPINK1 基因四个外显子中所有潜在编码 SNVs 的剪接效应。
我们的研究首先对 27 个先前使用 FLGSA 评估的 SPINK1 编码 SNVs 进行回顾性分析,然后对 35 个新的经过 FLGSA 测试的 SPINK1 编码 SNVs 进行前瞻性分析,接着进行数据外推,最后进行进一步验证。总共分析了 67 个 SPINK1 编码 SNVs,占 720 个可能编码 SNVs 的 9.3%。在这 67 个经过 FLGSA 分析的 SNVs 中,有 12 个被发现影响剪接。通过对 FLGSA 结果和 SpliceAI 预测的详细比较,我们推断 SPINK1 基因中其余 653 个未经测试的编码 SNVs 不太可能显著影响剪接。在 12 个影响剪接的事件中,有 9 个产生了正常剪接和异常剪接的转录本,而其余 3 个仅产生了异常剪接的转录本。这些影响剪接的 SNVs 仅存在于外显子 1 和 2 中,尤其是在外显子的第一个和/或最后一个编码核苷酸处。在这 12 个影响剪接的事件中,有 11 个是错义变异(占 506 个潜在错义变异的 2.17%),1 个是同义变异(占 164 个潜在同义变异的 0.61%)。值得注意的是,将 SpliceAI 截断值调整为 0.30 而不是传统的 0.20,在不降低敏感性的情况下可以提高特异性。
通过将 FLGSA 与 SpliceAI 相结合,我们确定 SPINK1 中所有可能的编码 SNVs 中不到 2%(1.67%)显著影响剪接结果。我们的研究结果强调了在研究基因的更广泛基因组序列背景下进行剪接分析的重要性,并突出了中间 SpliceAI 分数(0.20 到 0.80)所固有的不确定性。本研究通过前瞻性地解释与疾病相关基因中所有潜在的编码 SNVs,以高度的准确性代表了从外显子和基因组测序时代的回顾性分析向前瞻性分析转变的有意义尝试,为该领域做出了贡献。