The Mary Lyon Centre, MRC Harwell, Oxfordshire, United Kingdom.
Université de Strasbourg, CNRS, INSERM, Institut Clinique de la Souris (ICS), PHENOMIN, CELPHEDIA, Illkirch, France.
PLoS Genet. 2024 Mar 8;20(3):e1011187. doi: 10.1371/journal.pgen.1011187. eCollection 2024 Mar.
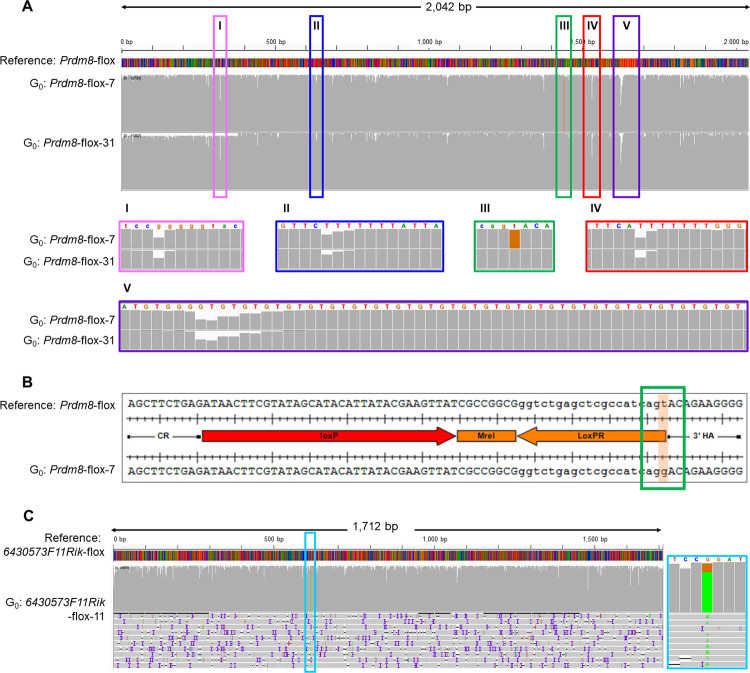
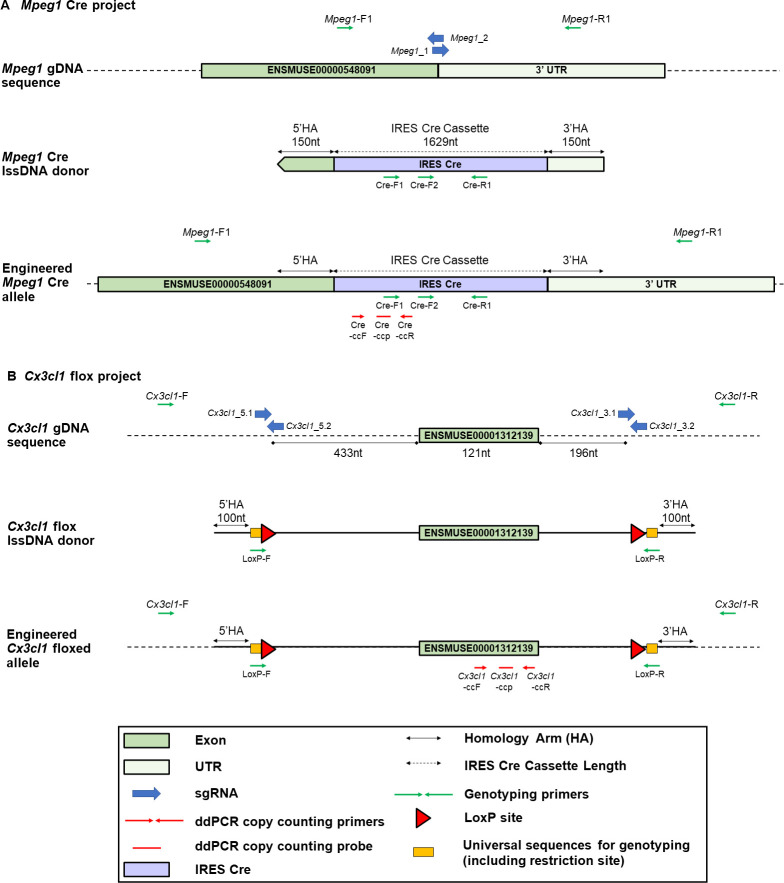
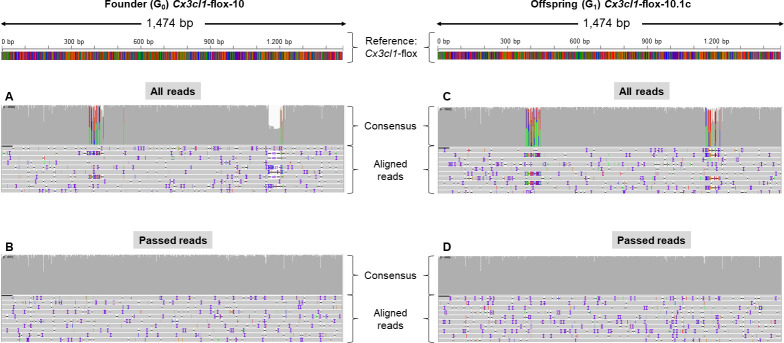
Recent developments in CRISPR/Cas9 genome-editing tools have facilitated the introduction of precise alleles, including genetic intervals spanning several kilobases, directly into the embryo. However, the introduction of donor templates, via homology directed repair, can be erroneous or incomplete and these techniques often produce mosaic founder animals. Thus, newly generated alleles must be verified at the sequence level across the targeted locus. Screening for the presence of the desired mutant allele using traditional sequencing methods can be challenging due to the size of the interval to be sequenced, together with the mosaic nature of founders.
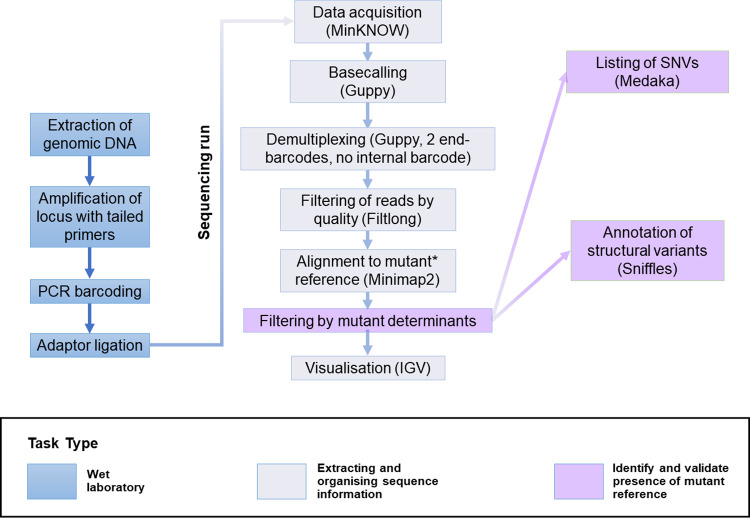
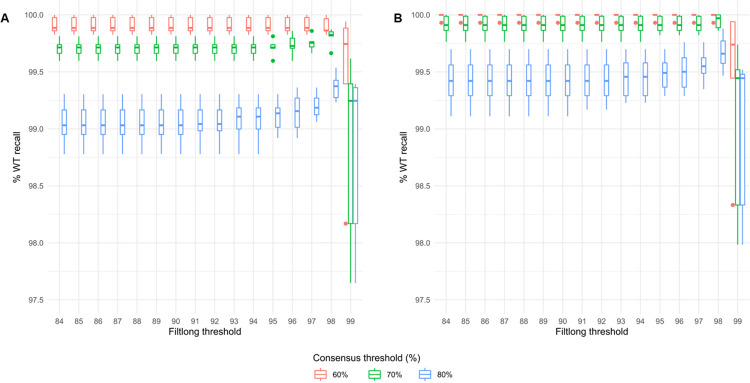
METHODOLOGY/PRINCIPAL FINDINGS: In order to help disentangle the genetic complexity of these animals, we tested the application of Oxford Nanopore Technologies long-read sequencing at the targeted locus and found that the achievable depth of sequencing is sufficient to offset the sequencing error rate associated with the technology used to validate targeted regions of interest. We have assembled an analysis workflow that facilitates interrogating the entire length of a targeted segment in a single read, to confirm that the intended mutant sequence is present in both heterozygous animals and mosaic founders. We used this workflow to compare the output of PCR-based and Cas9 capture-based targeted sequencing for validation of edited alleles.
Targeted long-read sequencing supports in-depth characterisation of all experimental models that aim to produce knock-in or conditional alleles, including those that contain a mix of genome-edited alleles. PCR- or Cas9 capture-based modalities bring different advantages to the analysis.
CRISPR/Cas9 基因组编辑工具的最新进展使得直接在胚胎中引入精确等位基因,包括跨越几个千碱基的遗传间隔,成为可能。然而,通过同源定向修复引入供体模板可能是错误的或不完整的,并且这些技术通常会产生嵌合体创始动物。因此,新生成的等位基因必须在目标基因座的序列水平上进行验证。由于要测序的间隔大小以及创始动物的嵌合性质,使用传统测序方法筛选所需突变等位基因的存在可能具有挑战性。
方法/主要发现:为了帮助理清这些动物的遗传复杂性,我们测试了在目标基因座应用 Oxford Nanopore Technologies 长读测序,并发现测序的可实现深度足以抵消用于验证目标感兴趣区域的技术相关的测序错误率。我们已经开发了一种分析工作流程,该流程可方便地在单个读取中询问目标片段的全长,以确认预期的突变序列存在于杂合动物和嵌合体创始动物中。我们使用该工作流程比较了基于 PCR 和 Cas9 捕获的靶向测序在验证编辑等位基因方面的输出。
靶向长读测序支持对所有旨在产生敲入或条件等位基因的实验模型进行深入表征,包括那些包含混合基因组编辑等位基因的模型。基于 PCR 或 Cas9 捕获的方法为分析带来了不同的优势。