Graduate Institute of Biomedical Informatics, College of Medical Science and Technology, Taipei Medical University, Taipei 11031, Taiwan.
Division of Pulmonary Medicine, Department of Internal Medicine, School of Medicine, College of Medicine, Taipei Medical University, Taipei 11031, Taiwan.
Genes (Basel). 2022 Apr 13;13(4):686. doi: 10.3390/genes13040686.
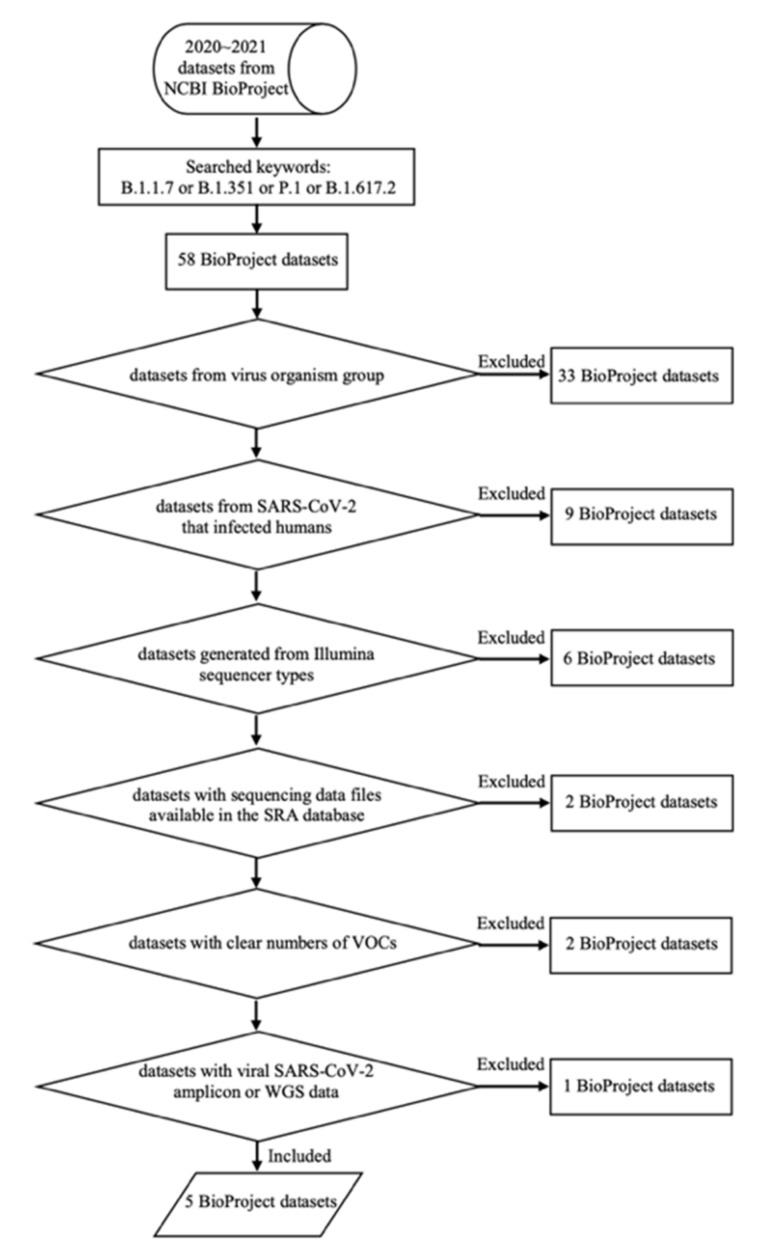
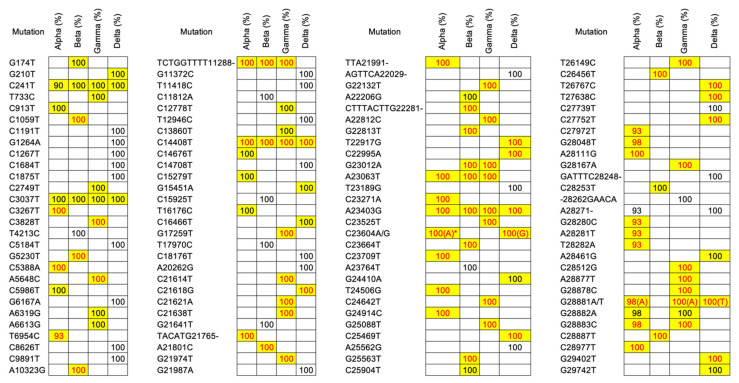
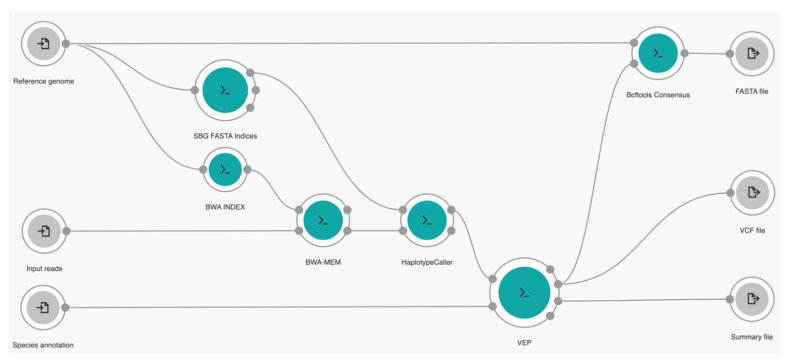
Several variants of the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) are emerging all over the world. Variant surveillance from genome sequencing has become crucial to determine if mutations in these variants are rendering the virus more infectious, potent, or resistant to existing vaccines and therapeutics. Meanwhile, analyzing many raw sequencing data repeatedly with currently available code-based bioinformatics tools is tremendously challenging to be implemented in this unprecedented pandemic time due to the fact of limited experts and computational resources. Therefore, in order to hasten variant surveillance efforts, we developed an installation-free cloud workflow for robust mutation profiling of SARS-CoV-2 variants from multiple Illumina sequencing data. Herein, 55 raw sequencing data representing four early SARS-CoV-2 variants of concern (Alpha, Beta, Gamma, and Delta) from an open-access database were used to test our workflow performance. As a result, our workflow could automatically identify mutated sites of the variants along with reliable annotation of the protein-coding genes at cost-effective and timely manner for all by harnessing parallel cloud computing in one execution under resource-limitation settings. In addition, our workflow can also generate a consensus genome sequence which can be shared with others in public data repositories to support global variant surveillance efforts.
多种新型严重急性呼吸综合征冠状病毒 2(SARS-CoV-2)变种正在全球范围内出现。通过基因组测序进行变种监测对于确定这些变种中的突变是否使病毒更具传染性、更强效或对现有疫苗和疗法产生抗性变得至关重要。与此同时,由于专家和计算资源有限,在这个前所未有的大流行时期,利用当前基于代码的生物信息学工具来重复分析大量原始测序数据在实施上极具挑战性。因此,为了加快变种监测工作,我们开发了一种免安装的云工作流程,用于从多个 Illumina 测序数据中对 SARS-CoV-2 变种进行强大的突变分析。在此,我们使用来自开放获取数据库的代表四种早期关注的 SARS-CoV-2 变种(Alpha、Beta、Gamma 和 Delta)的 55 个原始测序数据来测试我们的工作流程性能。结果表明,我们的工作流程可以在资源有限的设置下通过利用并行云计算在一次执行中自动识别变种的突变位点,并以具有成本效益且及时的方式对所有变体进行可靠的编码蛋白基因注释。此外,我们的工作流程还可以生成一个共识基因组序列,可在公共数据存储库中与他人共享,以支持全球变种监测工作。