Gravel Barbara, Renaux Alexandre, Papadimitriou Sofia, Smits Guillaume, Nowé Ann, Lenaerts Tom
Interuniversity Institute of Bioinformatics in Brussels, Université Libre de Bruxelles-Vrije Universiteit Brussel, 1050 Brussels, Belgium.
Department of Computer Science, Machine Learning Group, Université Libre de Bruxelles, 1050 Brussels, Belgium.
Bioinformatics. 2024 Mar 29;40(4). doi: 10.1093/bioinformatics/btae184.
Whole exome sequencing (WES) has emerged as a powerful tool for genetic research, enabling the collection of a tremendous amount of data about human genetic variation. However, properly identifying which variants are causative of a genetic disease remains an important challenge, often due to the number of variants that need to be screened. Expanding the screening to combinations of variants in two or more genes, as would be required under the oligogenic inheritance model, simply blows this problem out of proportion.
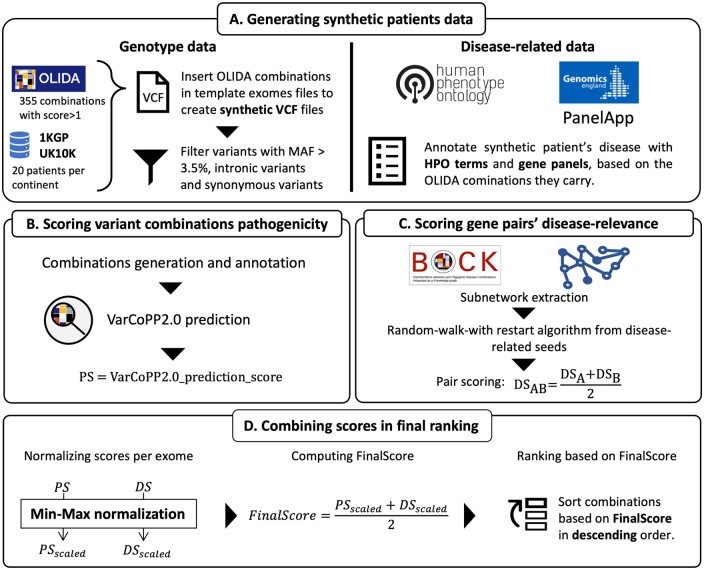
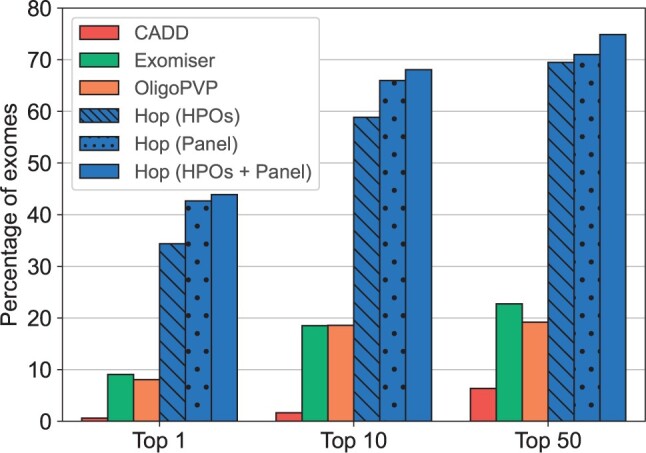
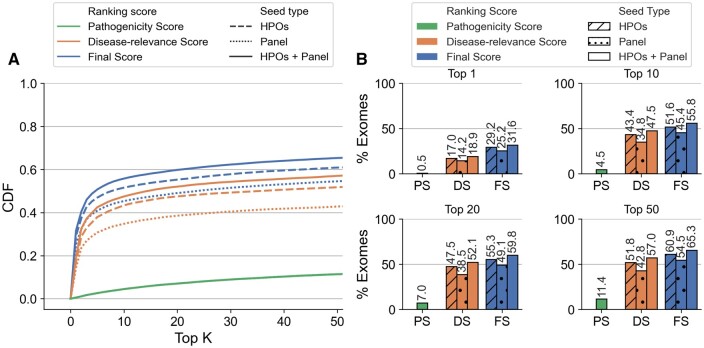
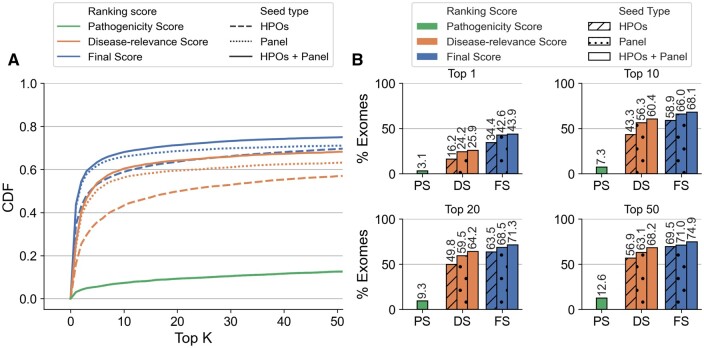
We present here the High-throughput oligogenic prioritizer (Hop), a novel prioritization method that uses direct oligogenic information at the variant, gene and gene pair level to detect digenic variant combinations in WES data. This method leverages information from a knowledge graph, together with specialized pathogenicity predictions in order to effectively rank variant combinations based on how likely they are to explain the patient's phenotype. The performance of Hop is evaluated in cross-validation on 36 120 synthetic exomes for training and 14 280 additional synthetic exomes for independent testing. Whereas the known pathogenic variant combinations are found in the top 20 in approximately 60% of the cross-validation exomes, 71% are found in the same ranking range when considering the independent set. These results provide a significant improvement over alternative approaches that depend simply on a monogenic assessment of pathogenicity, including early attempts for digenic ranking using monogenic pathogenicity scores.
Hop is available at https://github.com/oligogenic/HOP.
全外显子组测序(WES)已成为遗传研究的强大工具,能够收集大量有关人类遗传变异的数据。然而,正确识别哪些变异是导致遗传疾病的原因仍然是一项重大挑战,这通常是由于需要筛选的变异数量众多。将筛选扩展到两个或更多基因中的变异组合,就像在寡基因遗传模型下所要求的那样,只会使这个问题变得更加严重。
我们在此展示了高通量寡基因优先级排序器(Hop),这是一种新颖的优先级排序方法,它在变异、基因和基因对水平上使用直接的寡基因信息来检测WES数据中的双基因变异组合。该方法利用来自知识图谱的信息以及专门的致病性预测,以便根据变异组合解释患者表型的可能性有效地对其进行排名。在交叉验证中,对36120个用于训练的合成外显子组和另外14280个用于独立测试的合成外显子组评估了Hop的性能。虽然在大约60%的交叉验证外显子组中,已知的致病变异组合在前20名中被发现,但在考虑独立数据集时,71%的组合在相同的排名范围内被发现。这些结果相对于仅依赖单基因致病性评估的替代方法有了显著改进, 包括早期使用单基因致病性评分进行双基因排名的尝试。