Division of Epidemiology, Department of Medicine, Vanderbilt-Ingram Cancer Center, Vanderbilt Epidemiology Center, Vanderbilt University Medical Center, Nashville, TN, USA.
Department of Biomedical Informatics, Vanderbilt University School of Medicine, Nashville, TN, USA.
Nat Commun. 2024 Apr 26;15(1):3557. doi: 10.1038/s41467-024-47399-x.
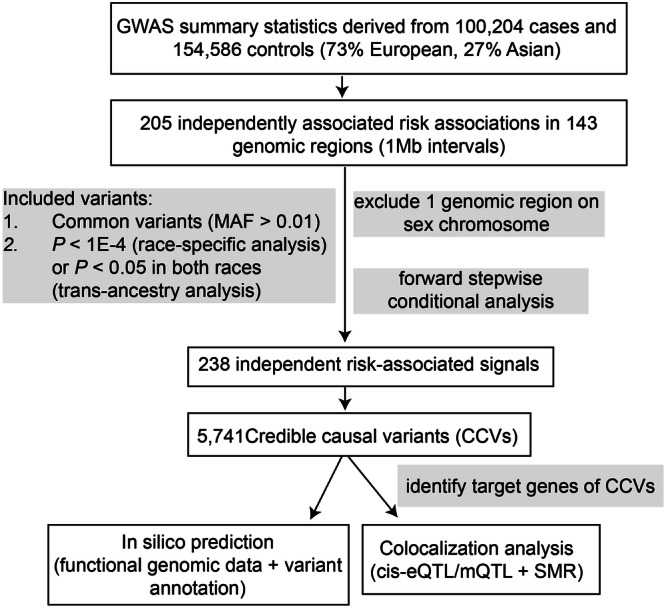
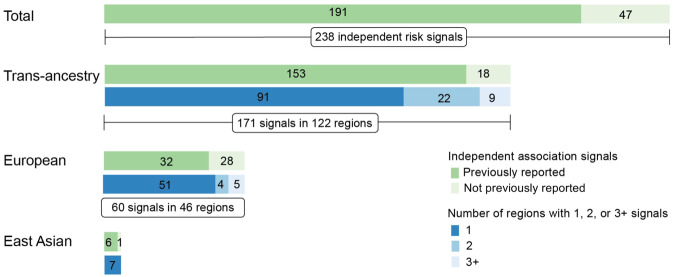
Genome-wide association studies (GWAS) have identified more than 200 common genetic variants independently associated with colorectal cancer (CRC) risk, but the causal variants and target genes are mostly unknown. We sought to fine-map all known CRC risk loci using GWAS data from 100,204 cases and 154,587 controls of East Asian and European ancestry. Our stepwise conditional analyses revealed 238 independent association signals of CRC risk, each with a set of credible causal variants (CCVs), of which 28 signals had a single CCV. Our cis-eQTL/mQTL and colocalization analyses using colorectal tissue-specific transcriptome and methylome data separately from 1299 and 321 individuals, along with functional genomic investigation, uncovered 136 putative CRC susceptibility genes, including 56 genes not previously reported. Analyses of single-cell RNA-seq data from colorectal tissues revealed 17 putative CRC susceptibility genes with distinct expression patterns in specific cell types. Analyses of whole exome sequencing data provided additional support for several target genes identified in this study as CRC susceptibility genes. Enrichment analyses of the 136 genes uncover pathways not previously linked to CRC risk. Our study substantially expanded association signals for CRC and provided additional insight into the biological mechanisms underlying CRC development.
全基因组关联研究(GWAS)已经独立鉴定出 200 多种与结直肠癌(CRC)风险相关的常见遗传变异,但因果变异和靶基因大多未知。我们试图使用东亚和欧洲血统的 100204 例病例和 154587 例对照的 GWAS 数据,对所有已知的 CRC 风险位点进行精细定位。我们的逐步条件分析揭示了 238 个独立的 CRC 风险关联信号,每个信号都有一组可信的因果变异(CCV),其中 28 个信号只有一个 CCV。我们使用来自 1299 人和 321 人的结直肠组织特异性转录组和甲基组数据进行 cis-eQTL/mQTL 和共定位分析,以及功能基因组研究,发现了 136 个潜在的 CRC 易感基因,包括 56 个以前未报道过的基因。对来自结直肠组织的单细胞 RNA-seq 数据的分析揭示了 17 个具有特定细胞类型中独特表达模式的潜在 CRC 易感基因。对全外显子组测序数据的分析为该研究中确定的几个作为 CRC 易感基因的靶基因提供了额外的支持。对 136 个基因的富集分析揭示了以前与 CRC 风险无关的途径。我们的研究大大扩展了 CRC 的关联信号,并为 CRC 发展的生物学机制提供了更多的见解。