Department of Mechanical and Aerospace Engineering, Jacobs School of Engineering, University of California San Diego, La Jolla CA 92093, United States of America.
Department of Biomedical Engineering, University of Virginia, Charlottesville, VA, United States of America; Robert M. Berne Cardiovascular Research Center, University of Virginia, Charlottesville, VA, United States of America.
Comput Biol Med. 2024 Jun;175:108499. doi: 10.1016/j.compbiomed.2024.108499. Epub 2024 Apr 24.
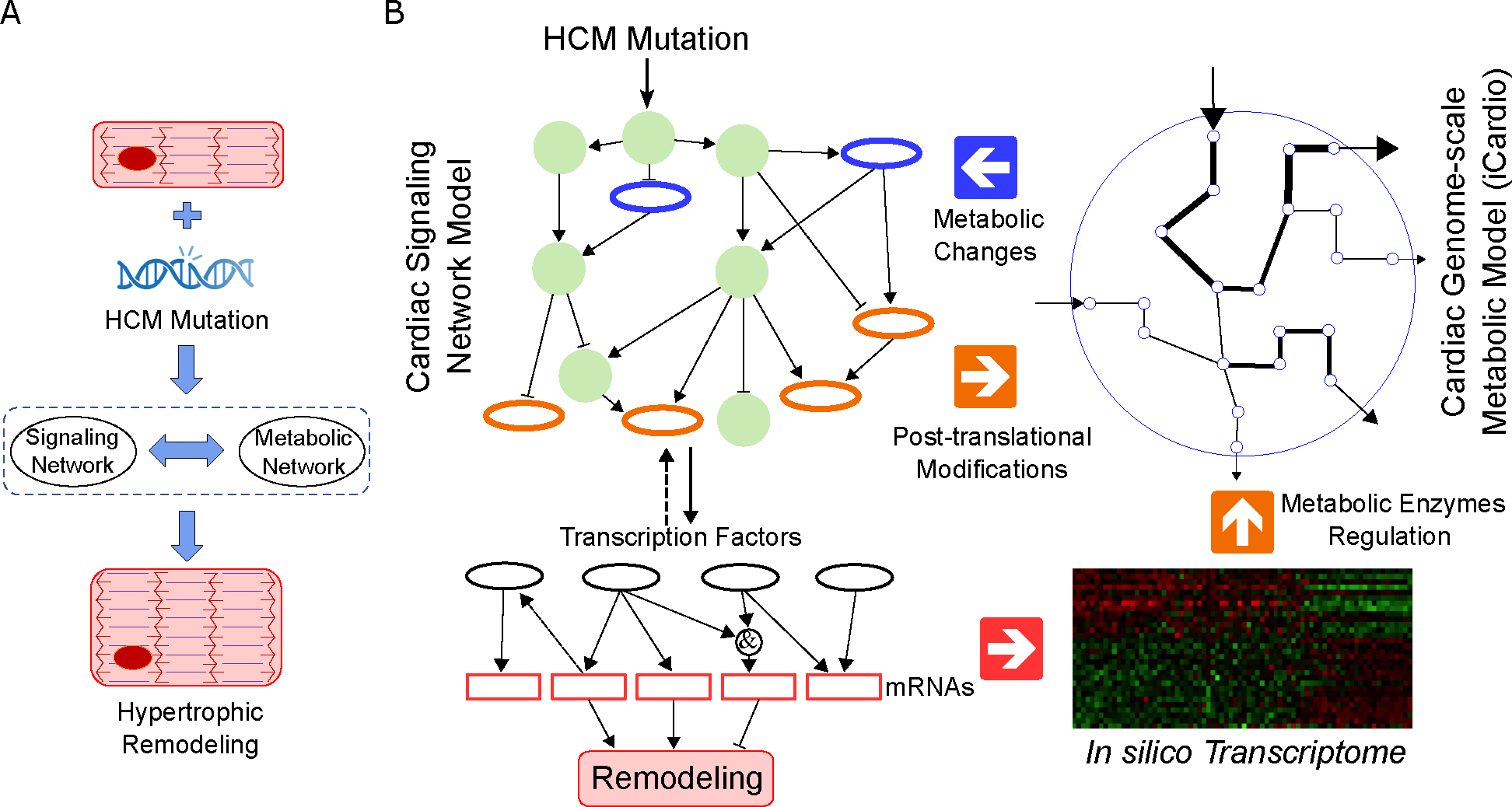
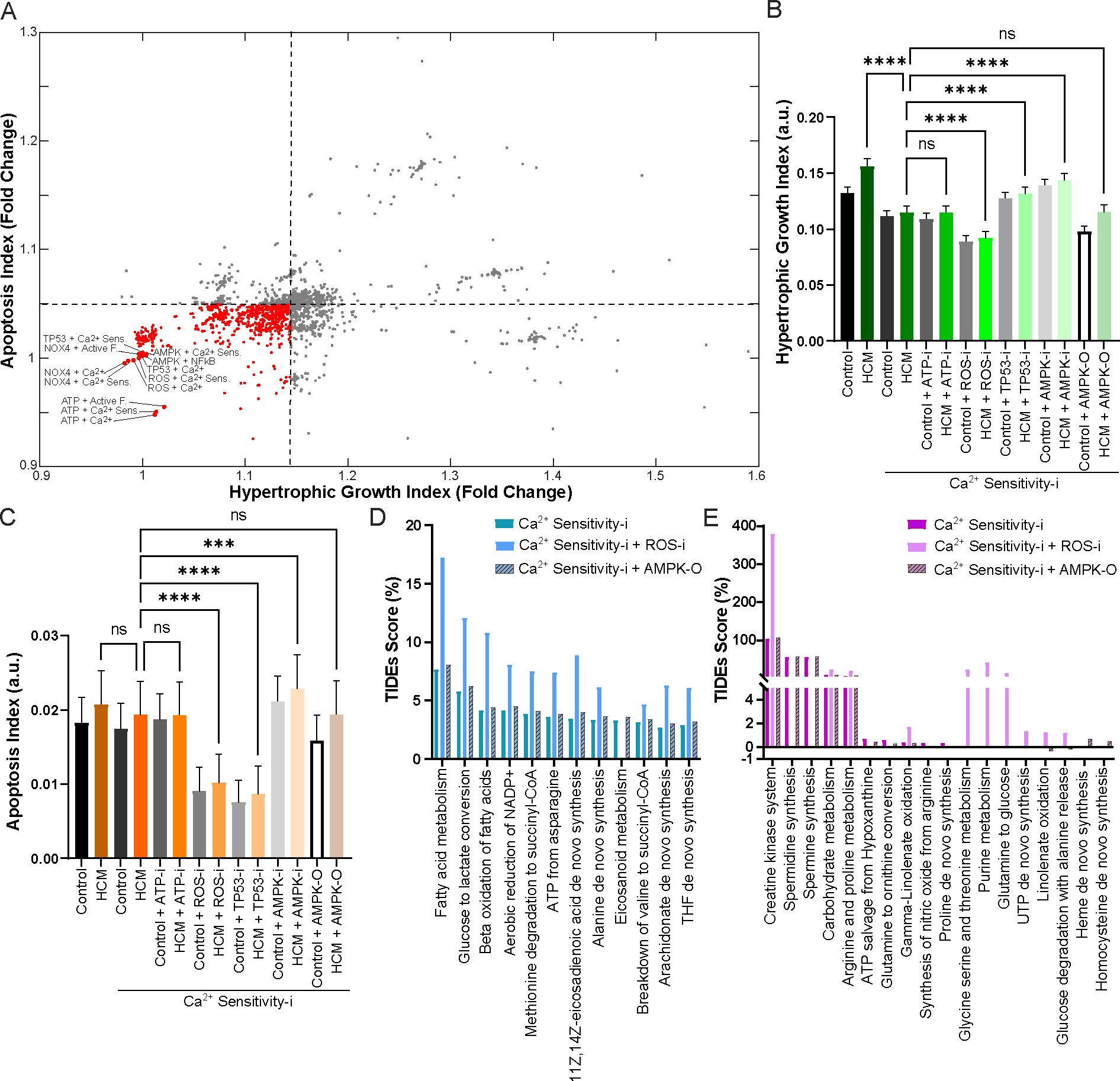
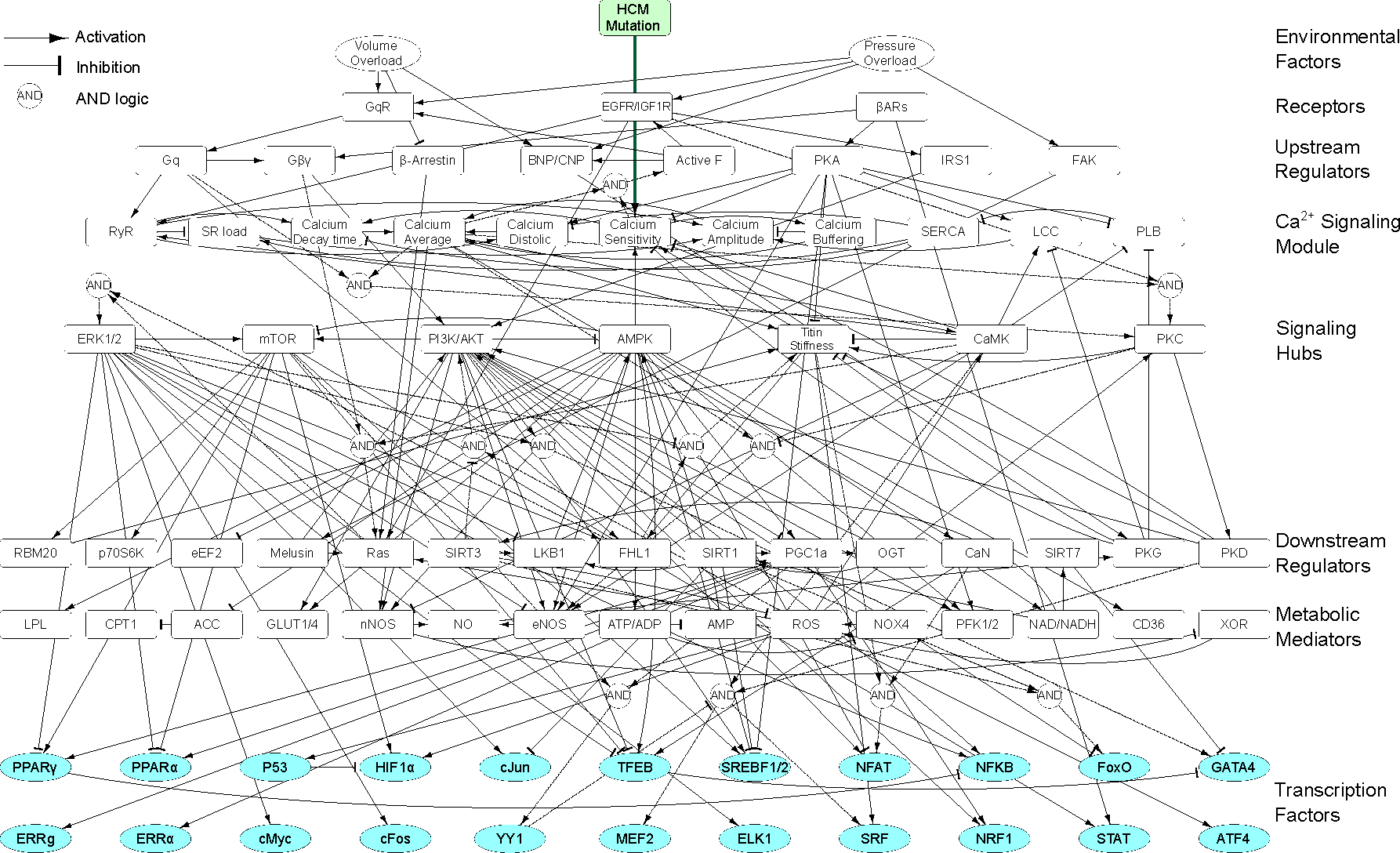
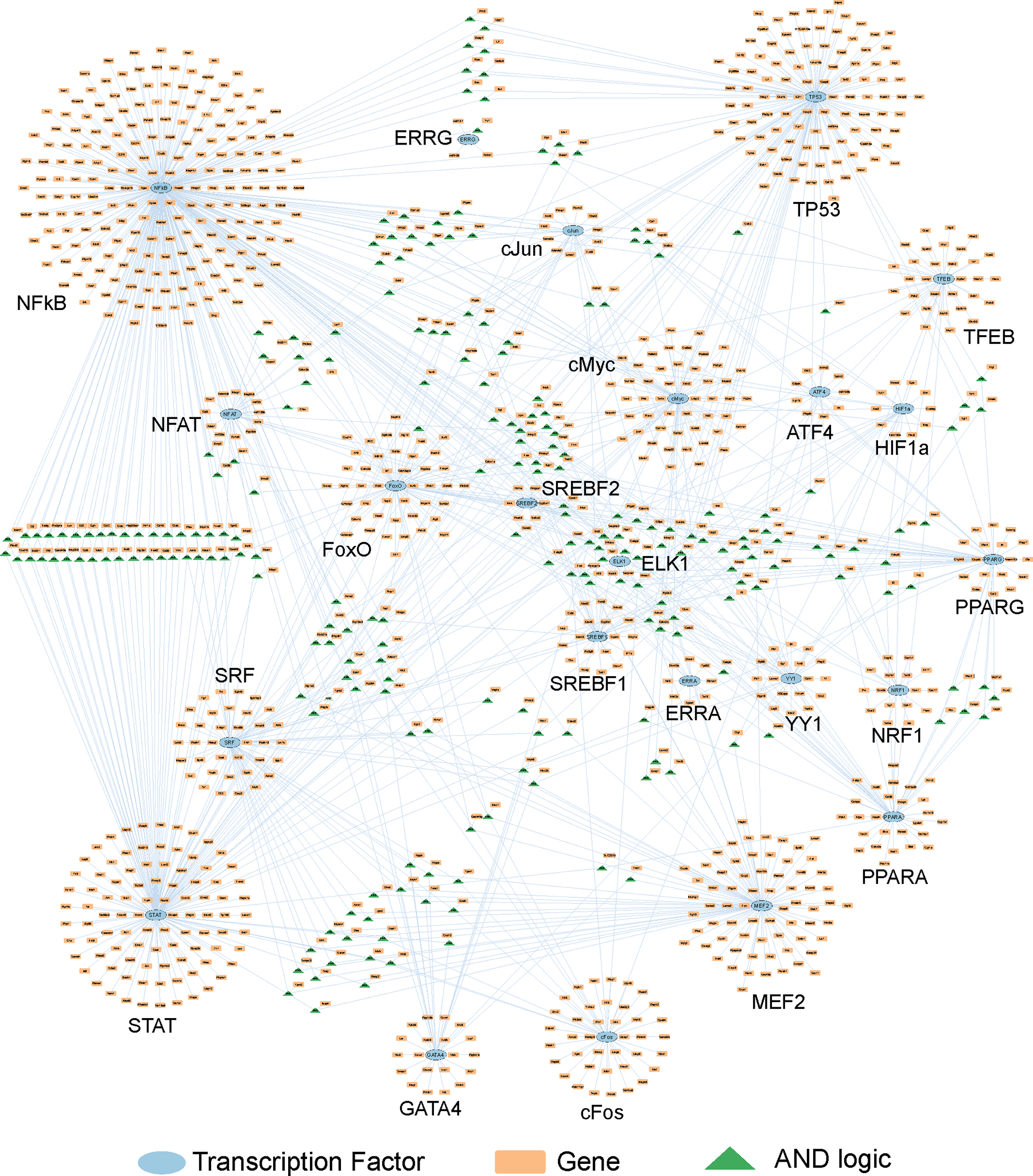
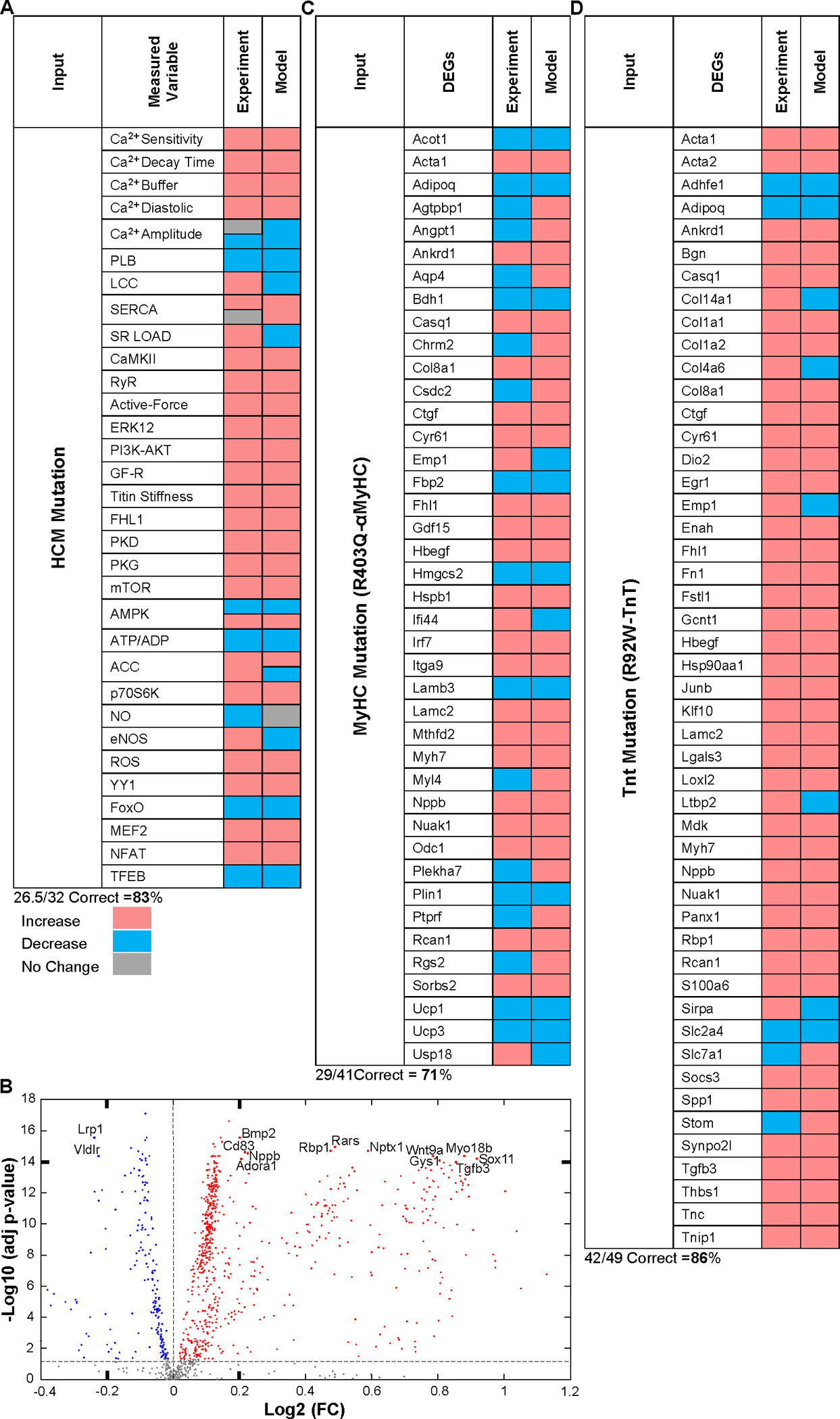
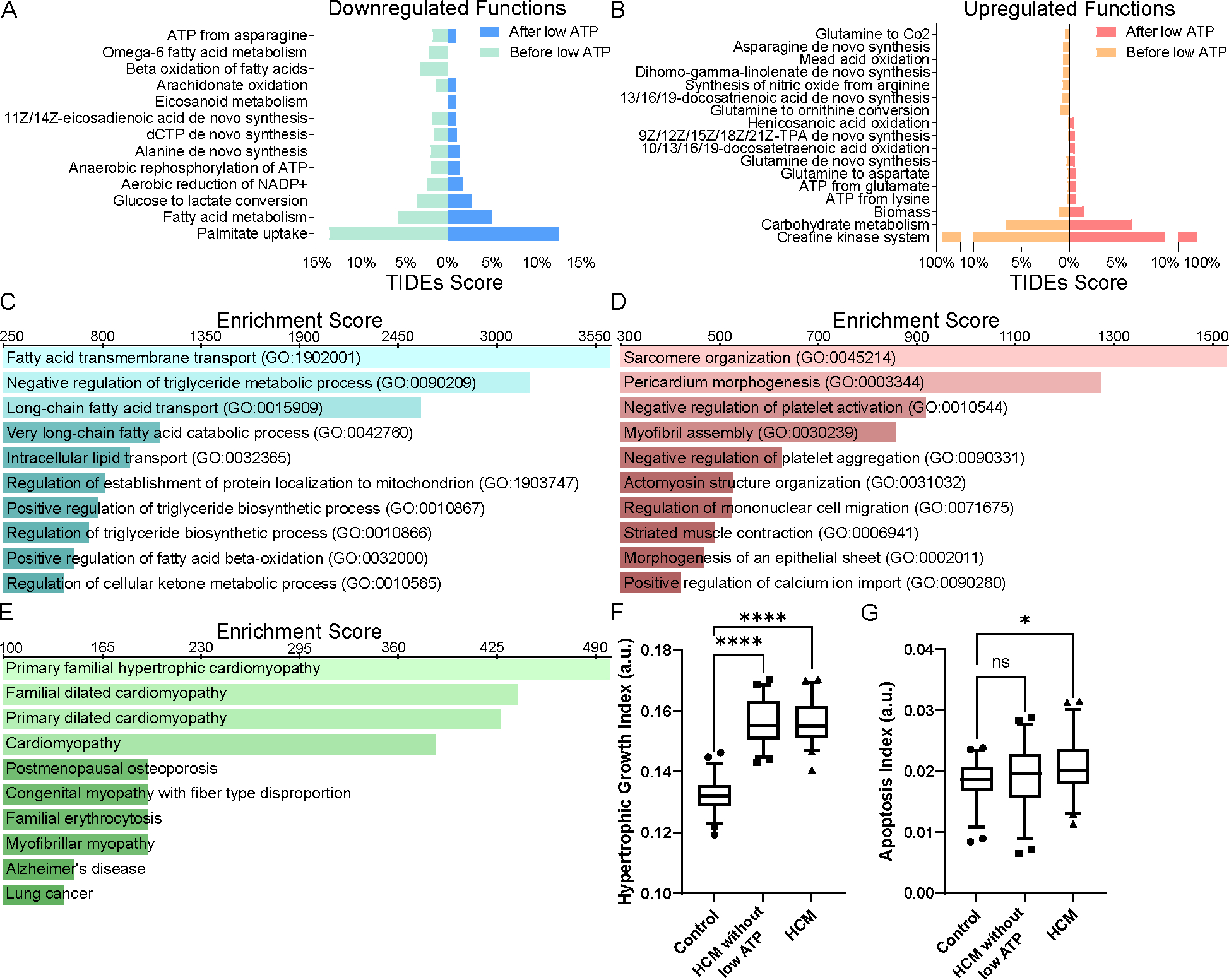
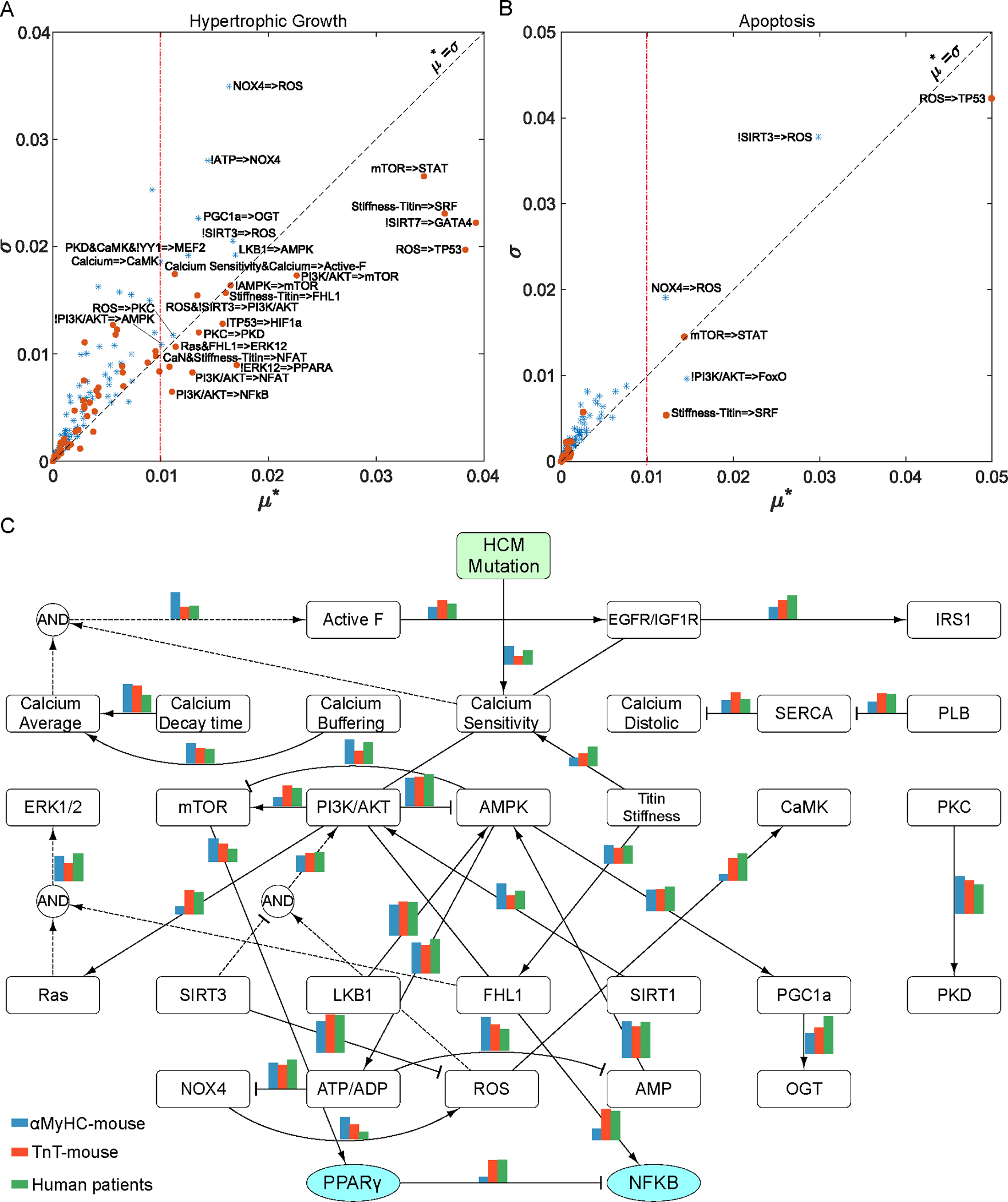
Familial hypertrophic cardiomyopathy (HCM) is a significant precursor of heart failure and sudden cardiac death, primarily caused by mutations in sarcomeric and structural proteins. Despite the extensive research on the HCM genotype, the complex and context-specific nature of many signaling and metabolic pathways linking the HCM genotype to phenotype has hindered therapeutic advancements for patients. Here, we have developed a computational model of HCM encompassing cardiomyocyte signaling and metabolic networks and their associated interactions. Utilizing a stochastic logic-based ODE approach, we linked cardiomyocyte signaling to the metabolic network through a gene regulatory network and post-translational modifications. We validated the model against published data on activities of signaling species in the HCM context and transcriptomes of two HCM mouse models (i.e., R403Q-αMyHC and R92W-TnT). Our model predicts that HCM mutation induces changes in metabolic functions such as ATP synthase deficiency and a transition from fatty acids to carbohydrate metabolism. The model indicated major shifts in glutamine-related metabolism and increased apoptosis after HCM-induced ATP synthase deficiency. We predicted that the transcription factors STAT, SRF, GATA4, TP53, and FoxO are the key regulators of cardiomyocyte hypertrophy and apoptosis in HCM in alignment with experiments. Moreover, we identified shared (e.g., activation of PGC1α by AMPK, and FHL1 by titin) and context-specific mechanisms (e.g., regulation of Ca2+ sensitivity by titin in HCM patients) that may control genotype-to-phenotype transition in HCM across different species or mutations. We also predicted potential combination drug targets for HCM (e.g., mavacamten plus ROS inhibitors) preventing or reversing HCM phenotype (i.e., hypertrophic growth, apoptosis, and metabolic remodeling) in cardiomyocytes. This study provides new insights into mechanisms linking genotype to phenotype in familial hypertrophic cardiomyopathy and offers a framework for assessing new treatments and exploring variations in HCM experimental models.
家族性肥厚型心肌病 (HCM) 是心力衰竭和心源性猝死的重要前兆,主要由肌节和结构蛋白的突变引起。尽管对 HCM 基因型进行了广泛的研究,但许多信号和代谢途径将 HCM 基因型与表型联系起来的复杂和特定于情境的性质阻碍了患者的治疗进展。在这里,我们开发了一个包含心肌细胞信号和代谢网络及其相关相互作用的 HCM 计算模型。我们利用基于随机逻辑的 ODE 方法,通过基因调控网络和翻译后修饰将心肌细胞信号与代谢网络联系起来。我们根据 HCM 背景下信号物种的活性和两种 HCM 小鼠模型(即 R403Q-αMyHC 和 R92W-TnT)的转录组数据对模型进行了验证。我们的模型预测,HCM 突变会导致代谢功能的变化,例如 ATP 合酶缺陷和从脂肪酸向碳水化合物代谢的转变。该模型表明,在 HCM 诱导的 ATP 合酶缺陷后,谷氨酰胺相关代谢和细胞凋亡会发生重大变化。我们预测转录因子 STAT、SRF、GATA4、TP53 和 FoxO 是 HCM 中心肌细胞肥大和细胞凋亡的关键调节剂,这与实验结果一致。此外,我们确定了共享(例如,AMPK 激活 PGC1α,titin 激活 FHL1)和特定于情境的机制(例如,titin 在 HCM 患者中调节 Ca2+敏感性),这些机制可能控制不同物种或突变中 HCM 的基因型到表型的转变。我们还预测了 HCM 的潜在联合药物靶点(例如,mavacamten 加 ROS 抑制剂),可防止或逆转心肌细胞中的 HCM 表型(即肥大生长、细胞凋亡和代谢重塑)。这项研究为家族性肥厚型心肌病中基因型与表型之间的联系提供了新的见解,并为评估新的治疗方法和探索 HCM 实验模型的变异性提供了框架。