McArdle Laboratory for Cancer Research, School of Medicine and Public Health, University of Wisconsin-Madison, Madison, Wisconsin, USA.
Department of Medical Microbiology and Immunology, School of Medicine and Public Health, University of Wisconsin-Madison, Madison, Wisconsin, USA.
mBio. 2024 Jun 12;15(6):e0093324. doi: 10.1128/mbio.00933-24. Epub 2024 May 14.
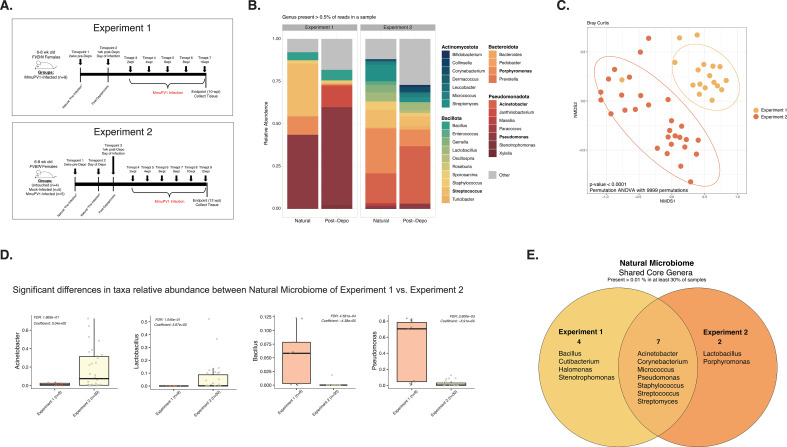
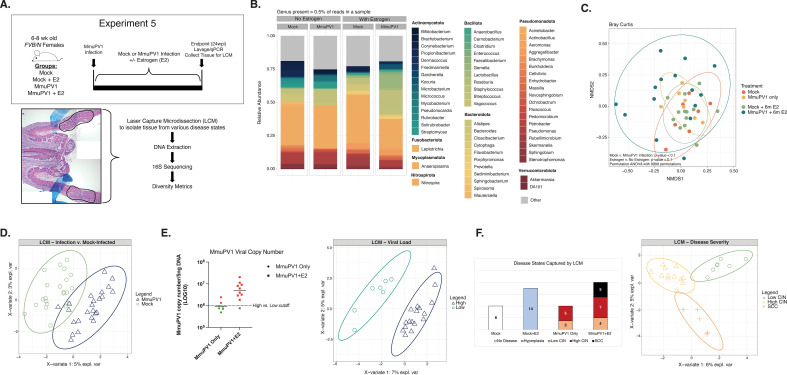
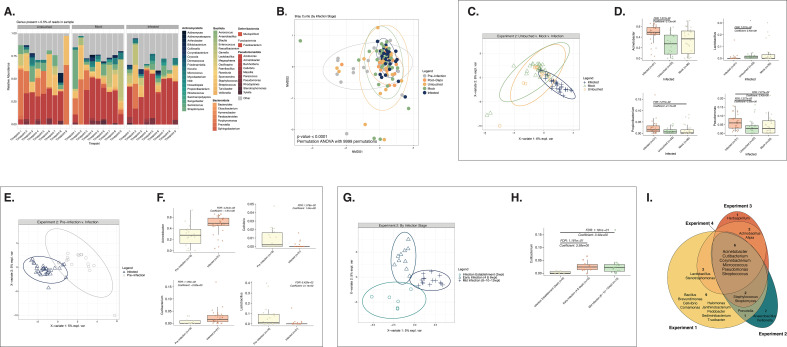
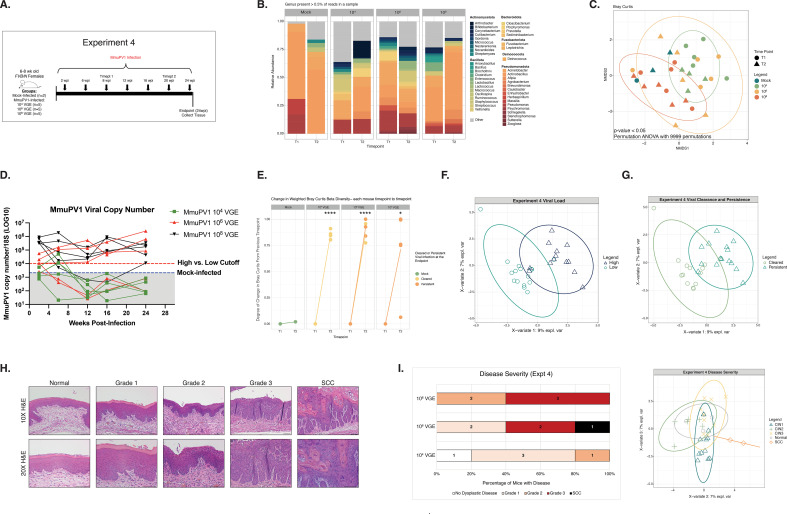
Human papillomaviruses (HPVs) are the most common sexually transmitted infection in the United States and are a major etiological agent of cancers in the anogenital tract and oral cavity. Growing evidence suggests changes in the host microbiome are associated with the natural history and ultimate outcome of HPV infection. We sought to define changes in the host cervicovaginal microbiome during papillomavirus infection, persistence, and pathogenesis using the murine papillomavirus (MmuPV1) cervicovaginal infection model. Cervicovaginal lavages were performed over a time course of MmuPV1 infection in immunocompetent female mice and extracted DNA was analyzed by qPCR to track MmuPV1 viral copy number. 16S ribosomal RNA (rRNA) gene sequencing was used to determine the composition and diversity of microbial communities throughout this time course. We also sought to determine whether specific microbial communities exist across the spectrum of MmuPV1-induced neoplastic disease. We, therefore, performed laser-capture microdissection to isolate regions of disease representing all stages of neoplastic disease progression (normal, low- and high-grade dysplasia, and cancer) from female reproductive tract tissue sections from MmuPV1-infected mice and performed 16S rRNA sequencing. Consistent with other studies, we found that the natural murine cervicovaginal microbiome is highly variable across different experiments. Despite these differences in initial microbiome composition between experiments, we observed that MmuPV1 persistence, viral load, and severity of disease influenced the composition of the cervicovaginal microbiome. These studies demonstrate that papillomavirus infection can alter the cervicovaginal microbiome.IMPORTANCEHuman papillomaviruses (HPVs) are the most common sexually transmitted infection in the United States. A subset of HPVs that infect the anogenital tract (cervix, vagina, anus) and oral cavity cause at least 5% of cancers worldwide. Recent evidence indicates that the community of microbial organisms present in the human cervix and vagina, known as the cervicovaginal microbiome, plays a role in HPV-induced cervical cancer. However, the mechanisms underlying this interplay are not well-defined. In this study, we infected the female reproductive tract of mice with a murine papillomavirus (MmuPV1) and found that key aspects of papillomavirus infection and disease influence the host cervicovaginal microbiome. This is the first study to define changes in the host microbiome associated with MmuPV1 infection in a preclinical animal model of HPV-induced cervical cancer. These results pave the way for using MmuPV1 infection models to further investigate the interactions between papillomaviruses and the host microbiome.
人乳头瘤病毒(HPV)是美国最常见的性传播感染,也是肛门生殖器和口腔癌症的主要病因。越来越多的证据表明,宿主微生物组的变化与 HPV 感染的自然史和最终结果有关。我们试图使用小鼠乳头瘤病毒(MmuPV1)宫颈阴道感染模型来定义 HPV 感染、持续存在和发病机制过程中宿主宫颈阴道微生物组的变化。在免疫功能正常的雌性小鼠的 MmuPV1 感染过程中进行了宫颈阴道灌洗,并通过 qPCR 分析提取的 DNA 以跟踪 MmuPV1 病毒拷贝数。16S 核糖体 RNA(rRNA)基因测序用于确定整个时间过程中微生物群落的组成和多样性。我们还试图确定在 MmuPV1 诱导的肿瘤疾病谱中是否存在特定的微生物群落。因此,我们进行了激光捕获显微切割,以从 MmuPV1 感染小鼠的生殖道组织切片中分离出代表肿瘤疾病进展各个阶段(正常、低级别和高级别发育不良以及癌症)的疾病区域,并进行了 16S rRNA 测序。与其他研究一致,我们发现天然的小鼠宫颈阴道微生物组在不同实验中变化很大。尽管这些实验之间的初始微生物组组成存在差异,但我们观察到 MmuPV1 的持续存在、病毒载量和疾病严重程度影响了宫颈阴道微生物组的组成。这些研究表明,乳头瘤病毒感染可以改变宫颈阴道微生物组。
人乳头瘤病毒(HPV)是美国最常见的性传播感染。感染肛门生殖器(宫颈、阴道、肛门)和口腔的 HPV 亚群导致全球至少 5%的癌症。最近的证据表明,存在于人类宫颈和阴道中的微生物群落,即宫颈阴道微生物组,在 HPV 诱导的宫颈癌中起作用。然而,这种相互作用的机制尚不清楚。在这项研究中,我们用一种小鼠乳头瘤病毒(MmuPV1)感染了雌性生殖道,并发现乳头瘤病毒感染和疾病的关键方面影响了宿主的宫颈阴道微生物组。这是第一项在 HPV 诱导的宫颈癌的临床前动物模型中定义与 MmuPV1 感染相关的宿主微生物组变化的研究。这些结果为使用 MmuPV1 感染模型进一步研究乳头瘤病毒与宿主微生物组之间的相互作用铺平了道路。