Laboratório de Biologia Molecular de Patógenos, Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil.
Departamento de Biofísica, Instituto de Biociências, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil.
Respir Res. 2024 May 18;25(1):211. doi: 10.1186/s12931-024-02835-w.
Bronchiectasis is a condition characterized by abnormal and irreversible bronchial dilation resulting from lung tissue damage and can be categorized into two main groups: cystic fibrosis (CF) and non-CF bronchiectasis (NCFB). Both diseases are marked by recurrent infections, inflammatory exacerbations, and lung damage. Given that infections are the primary drivers of disease progression, characterization of the respiratory microbiome can shed light on compositional alterations and susceptibility to antimicrobial drugs in these cases compared to healthy individuals.
To assess the microbiota in the two studied diseases, 35 subjects were recruited, comprising 10 NCFB and 13 CF patients and 12 healthy individuals. Nasopharyngeal swabs and induced sputum were collected, and total DNA was extracted. The DNA was then sequenced by the shotgun method and evaluated using the SqueezeMeta pipeline and R.
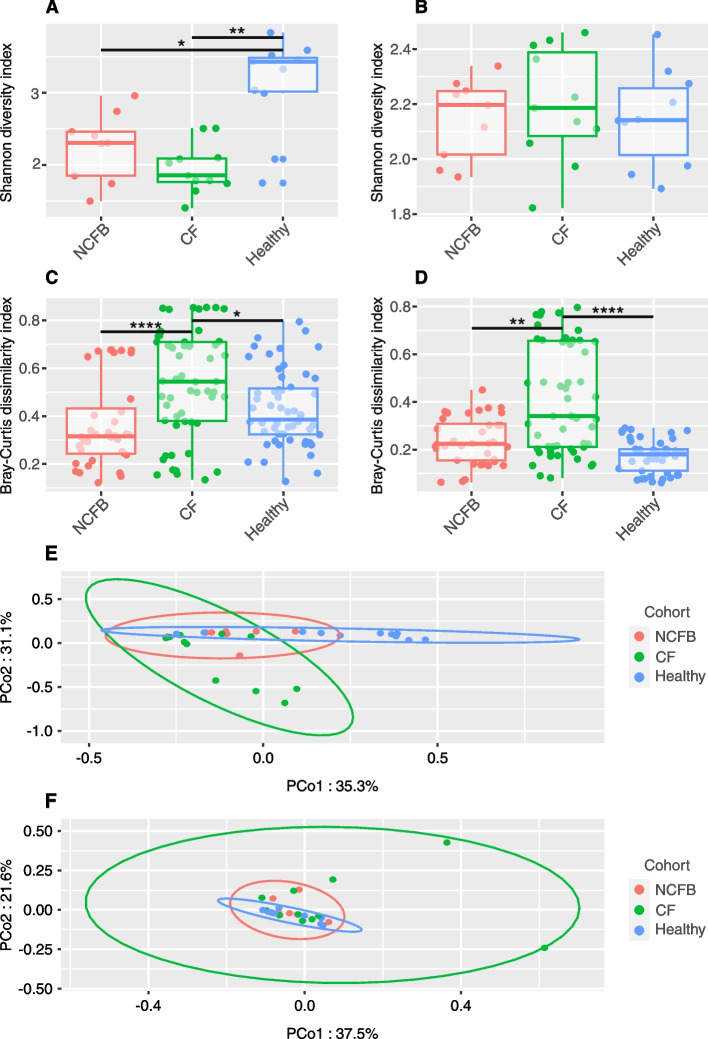
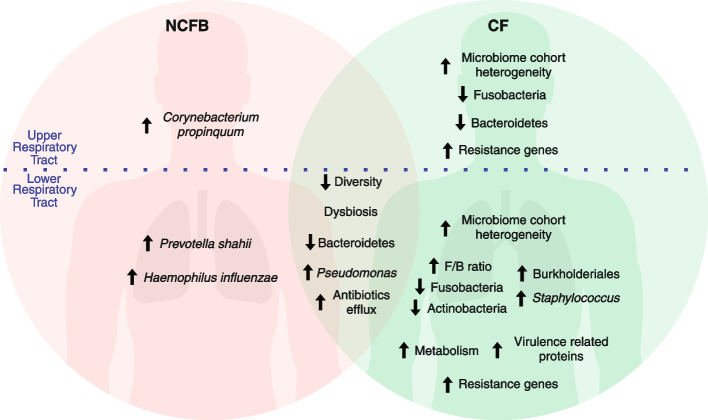
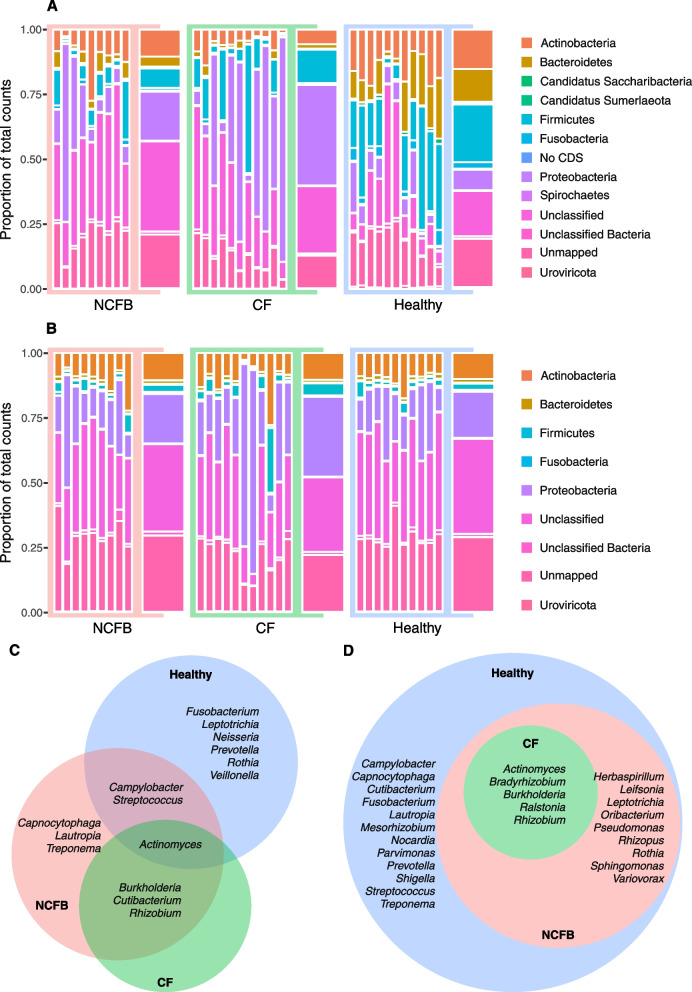
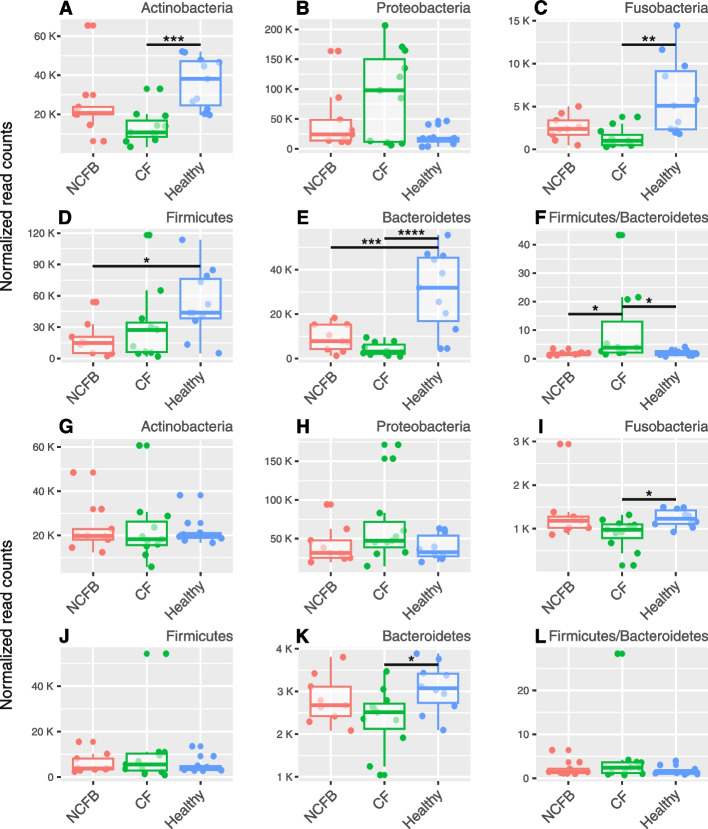
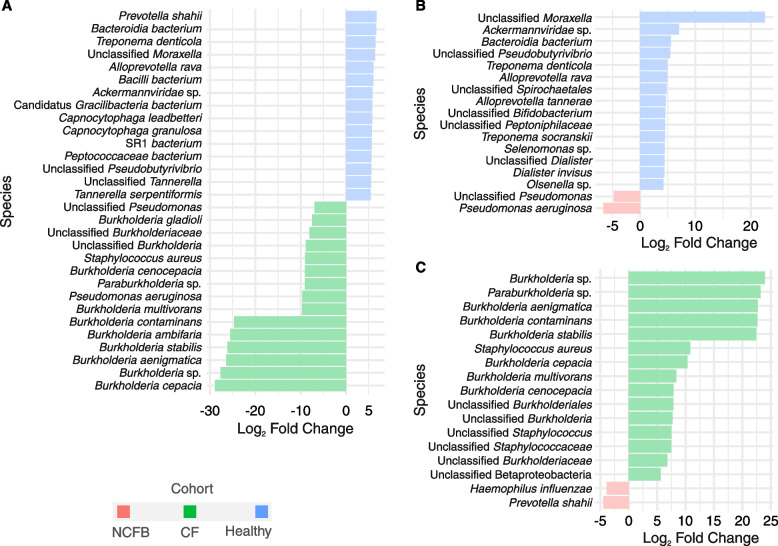
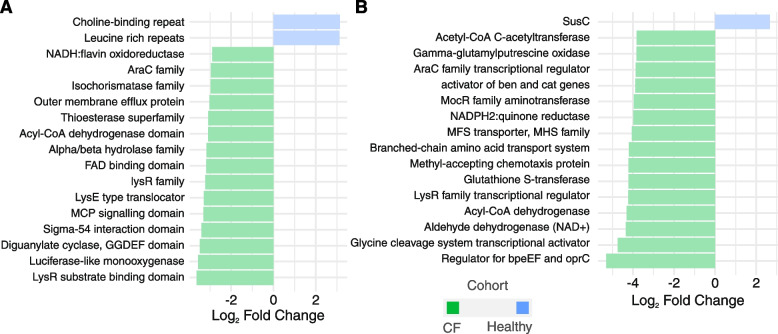
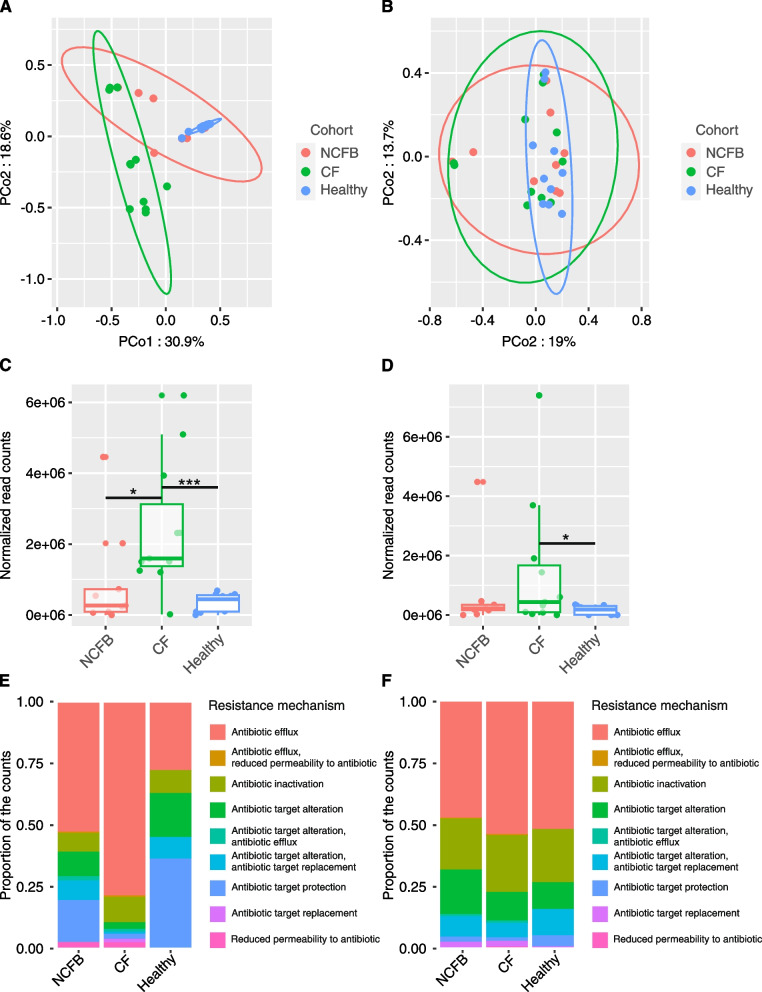
We observed reduced species diversity in both disease cohorts, along with distinct microbial compositions and profiles of antimicrobial resistance genes, compared to healthy individuals. The nasopharynx exhibited a consistent microbiota composition across all cohorts. Enrichment of members of the Burkholderiaceae family and an increased Firmicutes/Bacteroidetes ratio in the CF cohort emerged as key distinguishing factors compared to NCFB group. Staphylococcus aureus and Prevotella shahii also presented differential abundance in the CF and NCFB cohorts, respectively, in the lower respiratory tract. Considering antimicrobial resistance, a high number of genes related to antibiotic efflux were detected in both disease groups, which correlated with the patient's clinical data.
Bronchiectasis is associated with reduced microbial diversity and a shift in microbial and resistome composition compared to healthy subjects. Despite some similarities, CF and NCFB present significant differences in microbiome composition and antimicrobial resistance profiles, suggesting the need for customized management strategies for each disease.
支气管扩张症是一种由肺部组织损伤引起的异常且不可逆转的支气管扩张的疾病,可分为两个主要类型:囊性纤维化(CF)和非 CF 支气管扩张症(NCFB)。这两种疾病都以反复感染、炎症加重和肺部损伤为特征。由于感染是疾病进展的主要驱动因素,因此对呼吸微生物组进行特征分析可以揭示这些疾病与健康个体相比在组成变化和对抗生素药物的敏感性方面的差异。
为了评估两种研究疾病中的微生物组,招募了 35 名受试者,包括 10 名 NCFB 和 13 名 CF 患者和 12 名健康个体。采集鼻咽拭子和诱导痰,并提取总 DNA。然后通过鸟枪法对 DNA 进行测序,并使用 SqueezeMeta 管道和 R 进行评估。
我们观察到两个疾病队列的物种多样性均降低,与健康个体相比,微生物组成和抗菌药物耐药基因谱也存在明显差异。鼻咽部在所有队列中均表现出一致的微生物组成。与 NCFB 组相比,CF 组中伯克霍尔德氏菌科成员的丰度增加和厚壁菌门/拟杆菌门比值升高是关键的区分因素。金黄色葡萄球菌和普雷沃氏菌 Shahii 在下呼吸道中分别在 CF 和 NCFB 队列中表现出差异丰度。考虑到抗菌药物耐药性,在两个疾病组中均检测到大量与抗生素外排相关的基因,这些基因与患者的临床数据相关。
与健康受试者相比,支气管扩张症与微生物多样性降低以及微生物和耐药组组成的变化相关。尽管存在一些相似之处,但 CF 和 NCFB 在微生物组组成和抗菌药物耐药谱方面存在显著差异,这表明需要为每种疾病制定定制的管理策略。