Department of Econometrics and Operations Research, Tilburg University, Tilburg 5037AB, The Netherlands.
Department of Neurology, University Medical Center Utrecht, Utrecht 3584CX, The Netherlands.
Bioinformatics. 2024 Jun 3;40(6). doi: 10.1093/bioinformatics/btae326.
The completion of the genome has paved the way for genome-wide association studies (GWAS), which explained certain proportions of heritability. GWAS are not optimally suited to detect non-linear effects in disease risk, possibly hidden in non-additive interactions (epistasis). Alternative methods for epistasis detection using, e.g. deep neural networks (DNNs) are currently under active development. However, DNNs are constrained by finite computational resources, which can be rapidly depleted due to increasing complexity with the sheer size of the genome. Besides, the curse of dimensionality complicates the task of capturing meaningful genetic patterns for DNNs; therefore necessitates dimensionality reduction.
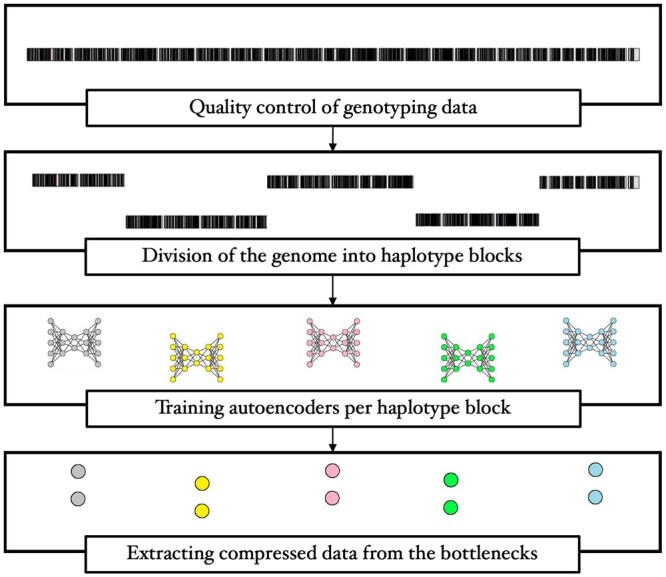
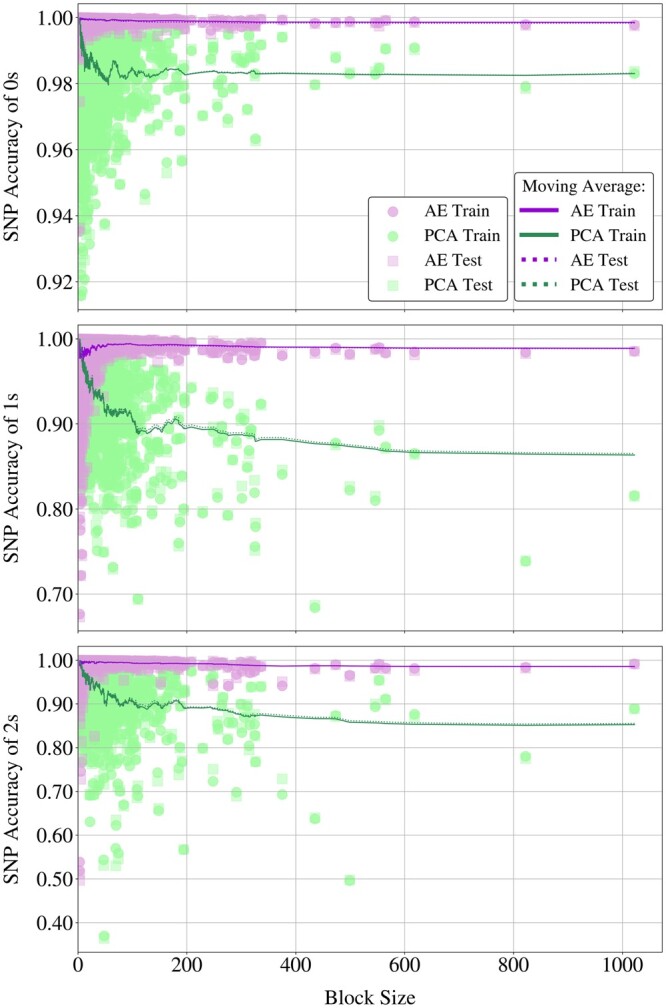
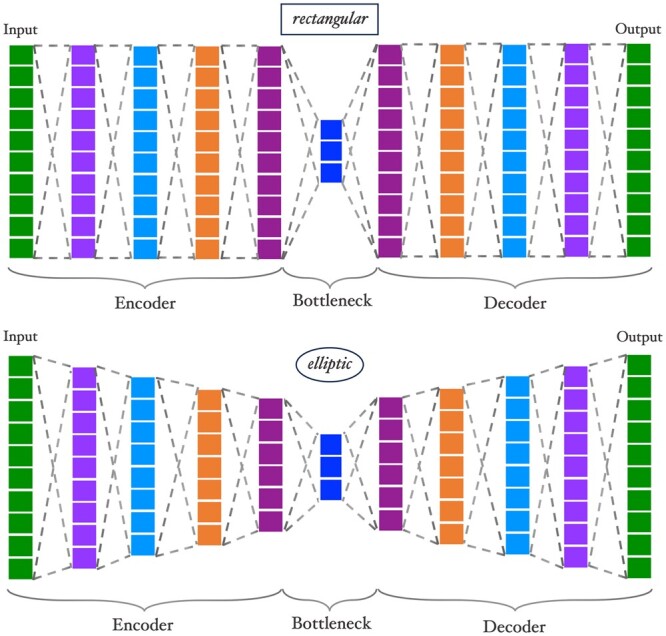
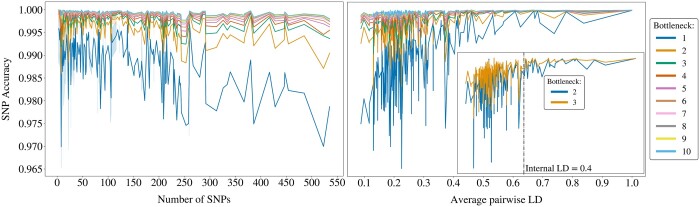
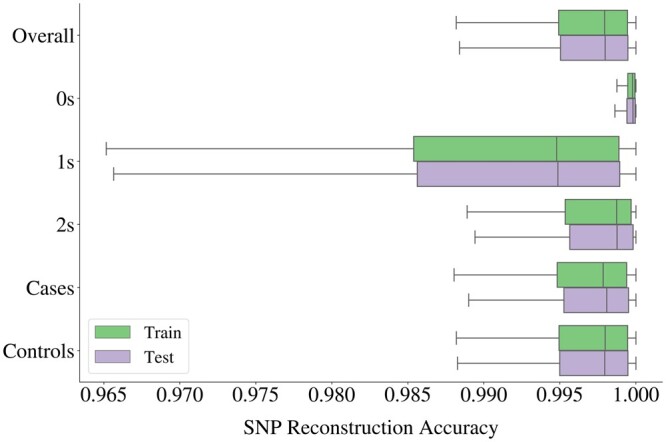
We propose a method to compress single nucleotide polymorphism (SNP) data, while leveraging the linkage disequilibrium (LD) structure and preserving potential epistasis. This method involves clustering correlated SNPs into haplotype blocks and training per-block autoencoders to learn a compressed representation of the block's genetic content. We provide an adjustable autoencoder design to accommodate diverse blocks and bypass extensive hyperparameter tuning. We applied this method to genotyping data from Project MinE, and achieved 99% average test reconstruction accuracy-i.e. minimal information loss-while compressing the input to nearly 10% of the original size. We demonstrate that haplotype-block based autoencoders outperform linear Principal Component Analysis (PCA) by approximately 3% chromosome-wide accuracy of reconstructed variants. To the extent of our knowledge, our approach is the first to simultaneously leverage haplotype structure and DNNs for dimensionality reduction of genetic data.
Data are available for academic use through Project MinE at https://www.projectmine.com/research/data-sharing/, contingent upon terms and requirements specified by the source studies. Code is available at https://github.com/gizem-tas/haploblock-autoencoders.
基因组的完成为全基因组关联研究(GWAS)铺平了道路,GWAS 解释了某些遗传率。GWAS 不太适合检测疾病风险中的非线性效应,这些效应可能隐藏在非加性相互作用(上位性)中。目前正在积极开发使用深度神经网络(DNN)等方法来检测上位性。然而,DNN 受到有限的计算资源的限制,由于基因组规模的巨大增加,这些资源可能会迅速耗尽。此外,维度的诅咒使 DNN 捕捉有意义的遗传模式的任务变得复杂;因此需要降维。
我们提出了一种压缩单核苷酸多态性(SNP)数据的方法,同时利用连锁不平衡(LD)结构并保留潜在的上位性。该方法涉及将相关的 SNP 聚类成单倍型块,并对每个块进行自动编码器训练,以学习块遗传内容的压缩表示。我们提供了一种可调节的自动编码器设计,以适应不同的块并避免广泛的超参数调整。我们将此方法应用于 Project MinE 的基因分型数据,实现了 99%的平均测试重建准确性-即最小信息丢失-同时将输入压缩到原始大小的近 10%。我们证明基于单倍型块的自动编码器比线性主成分分析(PCA)在重建变体的全染色体准确率上平均高出约 3%。据我们所知,我们的方法是第一个同时利用单倍型结构和 DNN 来降低遗传数据维度的方法。
数据可通过 Project MinE 在 https://www.projectmine.com/research/data-sharing/ 上供学术使用,前提是符合源研究规定的条款和要求。代码可在 https://github.com/gizem-tas/haploblock-autoencoders 上获得。