School of Medicine, Southeast University, Nanjing, China.
Chinese Academy of Medical Sciences Peking Union Medical College, Beijing, 100730, China.
J Transl Med. 2024 Jun 11;22(1):559. doi: 10.1186/s12967-024-05265-w.
Exploration of adaptive evolutionary changes at the genetic level in vaginal microbial communities during different stages of cervical cancer remains limited. This study aimed to elucidate the mutational profile of the vaginal microbiota throughout the progression of cervical disease and subsequently establish diagnostic models.
This study utilized a metagenomic dataset consisting of 151 subjects classified into four categories: invasive cervical cancer (CC) (n = 42), cervical intraepithelial neoplasia (CIN) (n = 43), HPV-infected (HPVi) patients without cervical lesions (n = 34), and healthy controls (n = 32). The analysis focused on changes in microbiome abundance and extracted information on genetic variation. Consequently, comprehensive multimodal microbial signatures associated with CC, encompassing taxonomic alterations, mutation signatures, and enriched metabolic functional pathways, were identified. Diagnostic models for predicting CC were established considering gene characteristics based on single nucleotide variants (SNVs).
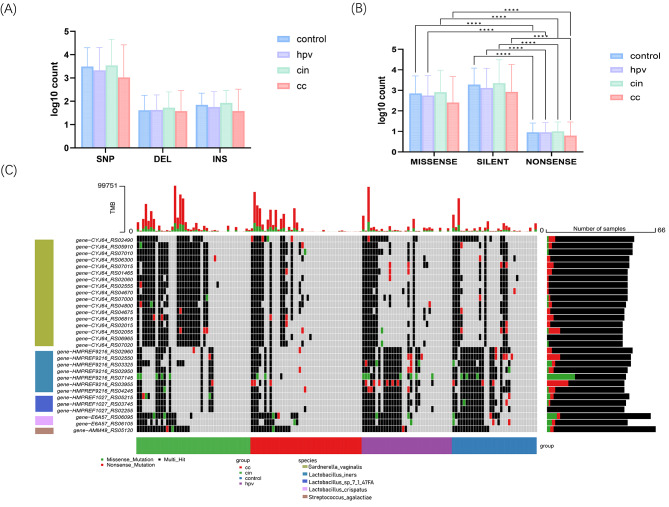
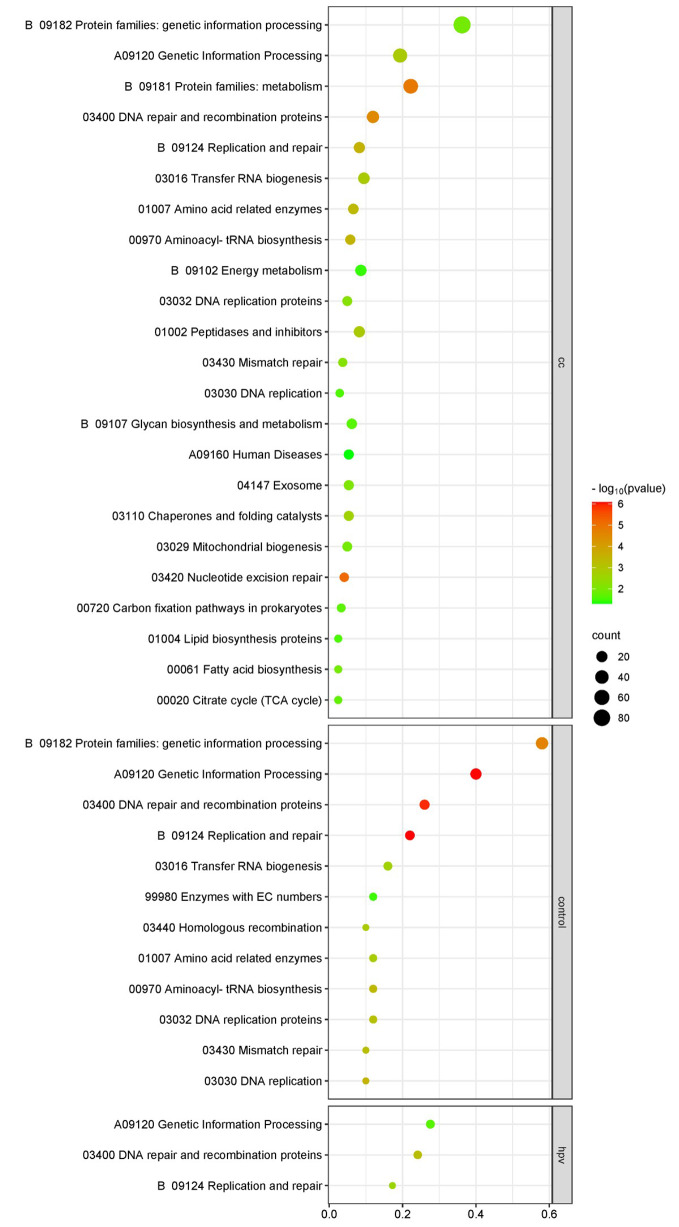
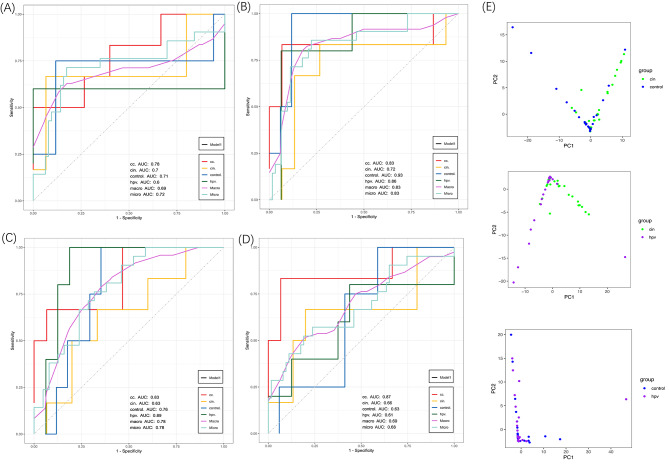
In this study, we screened and analyzed the abundances of 18 key microbial strains during CC progression. Additionally, 71,6358 non-redundant mutations were identified, predominantly consisting of SNVs that were further annotated into 25,773 genes. Altered abundances of SNVs and mutation types were observed across the four groups. Specifically, there were 9847 SNVs in the HPV-infected group and 14,892 in the CC group. Furthermore, two distinct mutation signatures corresponding to the benign and malignant groups were identified. The enriched metabolic pathways showed limited similarity with only two overlapping pathways among the four groups. HPVi patients exhibited active nucleotide biosynthesis, whereas patients with CC demonstrated a significantly higher abundance of signaling and cellular-associated protein families. In contrast, healthy controls showed a distinct enrichment in sugar metabolism. Moreover, biomarkers based on microbial SNV abundance displayed stronger diagnostic capability (cc.AUC = 0.87) than the species-level biomarkers (cc.AUC = 0.78). Ultimately, the integration of multimodal biomarkers demonstrated optimal performance for accurately identifying different cervical statuses (cc.AUC = 0.86), with an acceptable performance (AUC = 0.79) in the external testing set.
The vaginal microbiome exhibits specific SNV evolution in conjunction with the progression of CC, and serves as a specific biomarker for distinguishing between different statuses of cervical disease.
在宫颈癌的不同阶段,探索阴道微生物群落中遗传水平上的适应性进化变化仍然有限。本研究旨在阐明宫颈疾病进展过程中阴道微生物组的突变特征,并随后建立诊断模型。
本研究利用包含 151 个个体的宏基因组数据集,这些个体分为四组:浸润性宫颈癌(CC)(n=42)、宫颈上皮内瘤变(CIN)(n=43)、HPV 感染但无宫颈病变的患者(HPVi)(n=34)和健康对照(n=32)。分析重点是微生物组丰度的变化,并提取遗传变异信息。随后,确定了与 CC 相关的全面多模态微生物特征,包括分类学改变、突变特征和丰富的代谢功能途径。根据单核苷酸变异(SNV),考虑基因特征,建立了预测 CC 的诊断模型。
在本研究中,我们筛选和分析了 CC 进展过程中 18 种关键微生物菌株的丰度。此外,鉴定了 716358 个非冗余突变,主要由 SNV 组成,进一步注释为 25773 个基因。在四个组中观察到 SNV 和突变类型的丰度改变。具体而言,HPV 感染组中有 9847 个 SNV,CC 组中有 14892 个 SNV。此外,鉴定到与良性和恶性组相对应的两个独特的突变特征。富集的代谢途径与四个组中仅有两个重叠途径具有有限的相似性。HPV 感染患者表现出活跃的核苷酸合成,而 CC 患者表现出信号和细胞相关蛋白家族的显著更高丰度。相比之下,健康对照显示出独特的糖代谢富集。此外,基于微生物 SNV 丰度的生物标志物显示出比种属水平生物标志物更强的诊断能力(cc.AUC=0.87)。最终,多模态生物标志物的整合表现出准确识别不同宫颈状态的最佳性能(cc.AUC=0.86),在外部测试集中具有可接受的性能(AUC=0.79)。
阴道微生物组在 CC 进展过程中表现出特定的 SNV 进化,并且是区分不同宫颈疾病状态的特异性生物标志物。