Zhang Yiqun, Chen Fengju, Balic Marija, Creighton Chad J
Dan L. Duncan Comprehensive Cancer Center, Baylor College of Medicine, One Baylor Plaza, MS305, Houston, TX, 77030, USA.
Division of Oncology, Department of Internal Medicine, Medical University of Graz, Graz, Austria.
Breast Cancer Res. 2024 Jun 12;26(1):98. doi: 10.1186/s13058-024-01855-0.
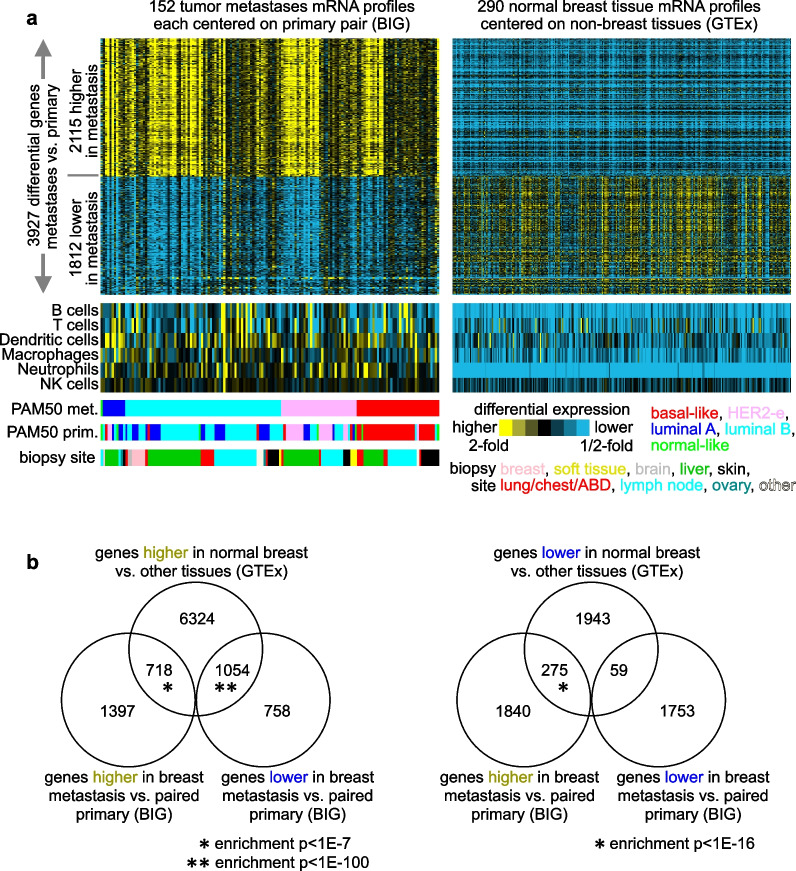
The differential gene expression profile of metastatic versus primary breast tumors represents an avenue for discovering new or underappreciated pathways underscoring processes of metastasis. However, as tumor biopsy samples are a mixture of cancer and non-cancer cells, most differentially expressed genes in metastases would represent confounders involving sample biopsy site rather than cancer cell biology.
By paired analysis, we defined a top set of differentially expressed genes in breast cancer metastasis versus primary tumors using an RNA-sequencing dataset of 152 patients from The Breast International Group Aiming to Understand the Molecular Aberrations dataset (BIG-AURORA). To filter the genes higher in metastasis for genes essential for breast cancer proliferation, we incorporated CRISPR-based data from breast cancer cell lines.
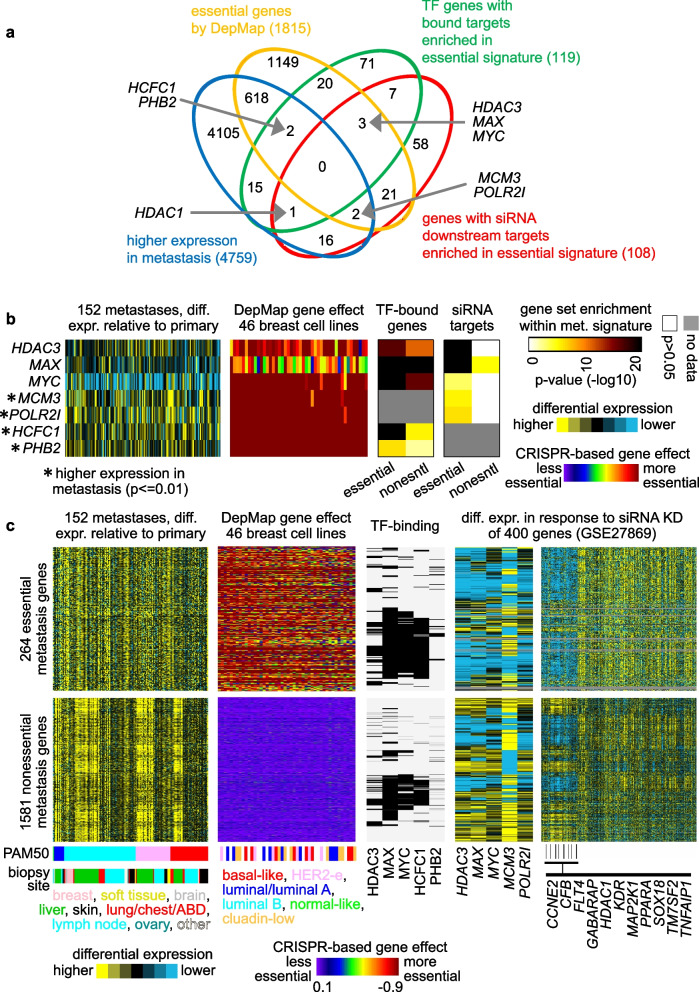
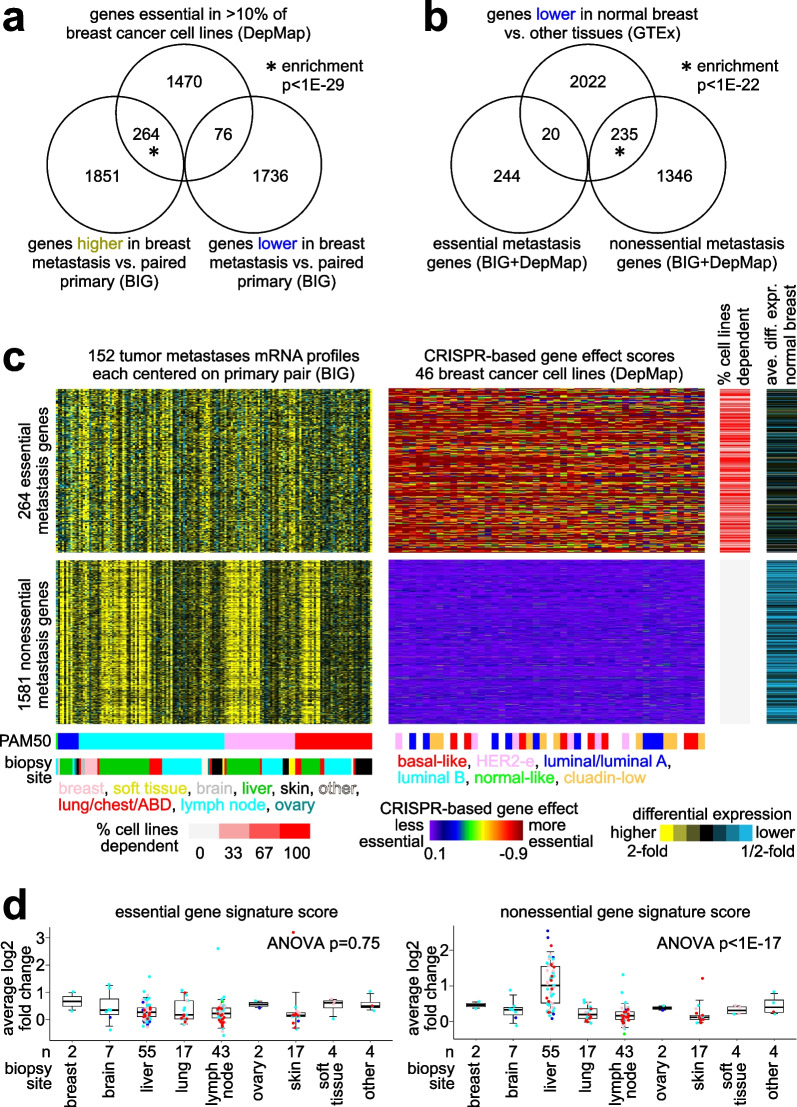
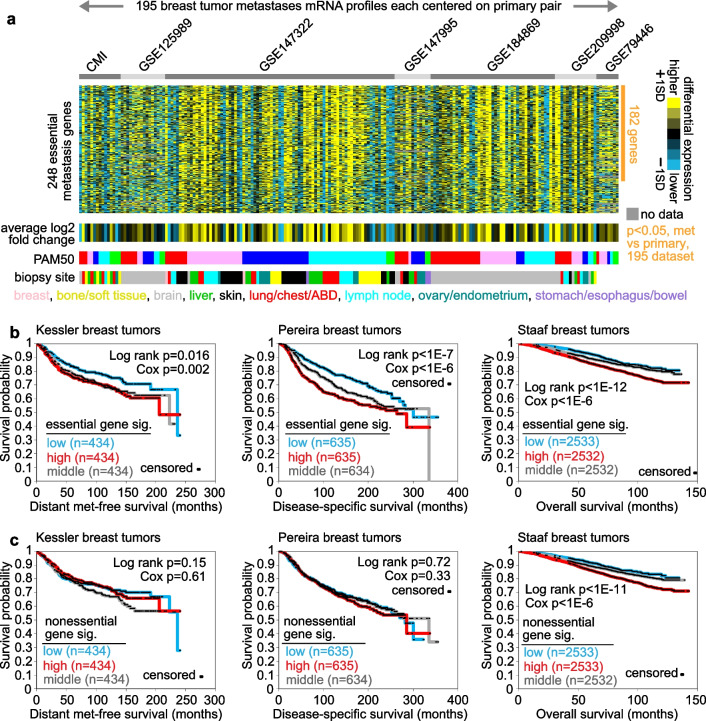
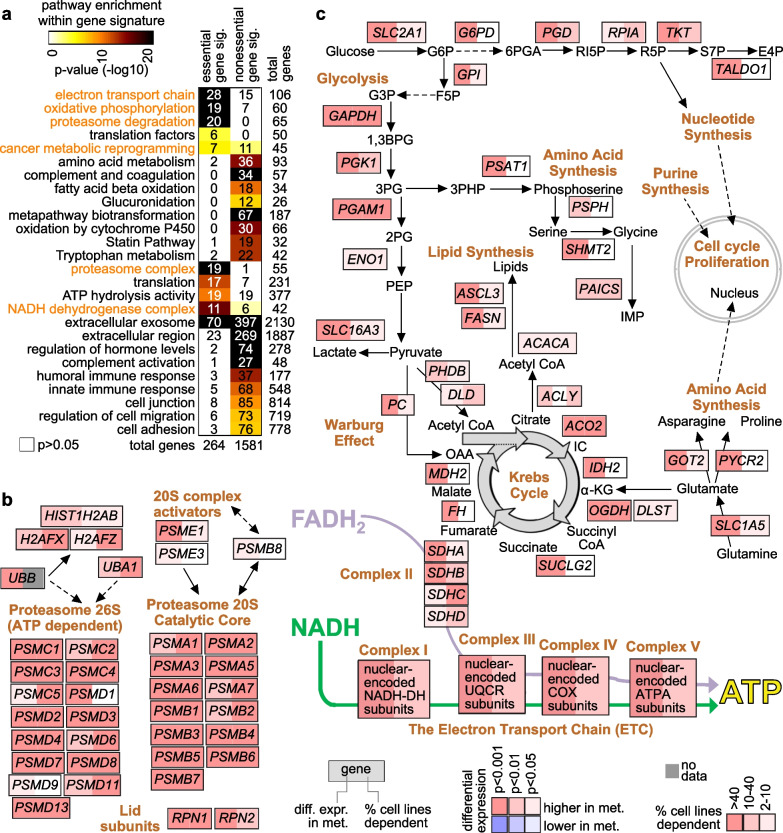
A significant fraction of genes with higher expression in metastasis versus paired primary were essential by CRISPR. These 264 genes represented an essential signature of breast cancer metastasis. In contrast, nonessential metastasis genes largely involved tumor biopsy site. The essential signature predicted breast cancer patient outcome based on primary tumor expression patterns. Pathways underlying the essential signature included proteasome degradation, the electron transport chain, oxidative phosphorylation, and cancer metabolic reprogramming. Transcription factors MYC, MAX, HDAC3, and HCFC1 each bound significant fractions of essential genes.
Associations involving the essential gene signature of breast cancer metastasis indicate true biological changes intrinsic to cancer cells, with important implications for applying existing therapies or developing alternate therapeutic approaches.
转移性乳腺癌与原发性乳腺癌的差异基因表达谱是发现新的或未被充分认识的转移途径的一条途径。然而,由于肿瘤活检样本是癌细胞和非癌细胞的混合物,转移灶中大多数差异表达基因可能代表涉及样本活检部位的混杂因素,而非癌细胞生物学因素。
通过配对分析,我们利用来自旨在了解分子畸变数据集(BIG - AURORA)的152例患者的RNA测序数据集,确定了一组乳腺癌转移灶与原发肿瘤中差异表达的顶级基因。为了从乳腺癌增殖所必需的基因中筛选出转移灶中表达较高的基因,我们纳入了来自乳腺癌细胞系的基于CRISPR的数据。
与配对的原发肿瘤相比,转移灶中表达较高的基因中有很大一部分通过CRISPR验证是必需的。这264个基因代表了乳腺癌转移的一个必需特征。相比之下,非必需的转移基因主要涉及肿瘤活检部位。基于原发肿瘤的表达模式,这个必需特征可预测乳腺癌患者的预后。该必需特征所涉及的途径包括蛋白酶体降解、电子传递链、氧化磷酸化和癌症代谢重编程。转录因子MYC、MAX、HDAC3和HCFC1分别与很大一部分必需基因结合。
涉及乳腺癌转移必需基因特征的关联表明癌细胞内在的真实生物学变化,这对于应用现有疗法或开发替代治疗方法具有重要意义。