Gokhman David, Harris Keith D, Carmi Shai, Greenbaum Gili
Department of Molecular Genetics, The Weizmann Institute of Science, Rehovot 76100, Israel.
Department of Ecology, Evolution and Behavior, The Hebrew University of Jerusalem, Jerusalem 91904, Israel.
bioRxiv. 2024 Jun 6:2024.02.22.581566. doi: 10.1101/2024.02.22.581566.
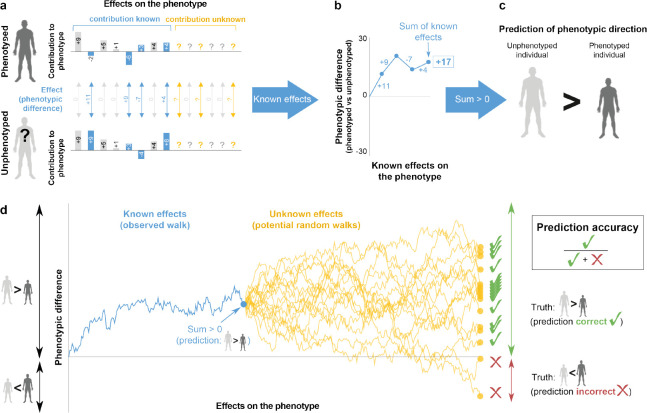
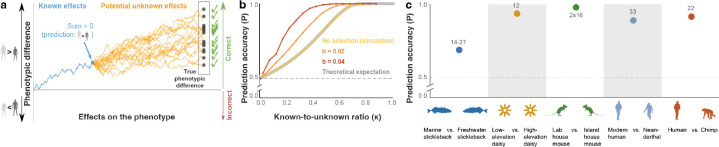
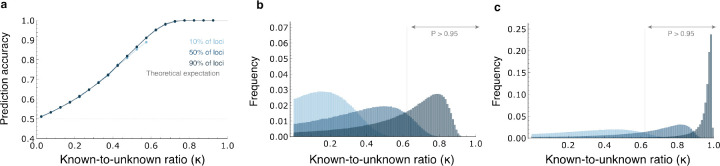
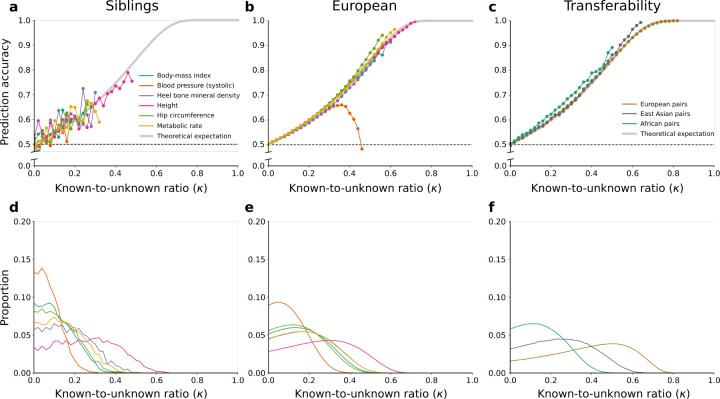
Predicting phenotypes from genomic data is a key goal in genetics, but for most complex phenotypes, predictions are hampered by incomplete genotype-to-phenotype mapping. Here, we describe a more attainable approach than quantitative predictions, which is aimed at qualitatively predicting phenotypic differences. Despite incomplete genotype-to-phenotype mapping, we show that it is relatively easy to determine which of two individuals has a greater phenotypic value. This question is central in many scenarios, e.g., comparing disease risk between individuals, the yield of crop strains, or the anatomy of extinct vs extant species. To evaluate prediction accuracy, i.e., the probability that the individual with the greater predicted phenotype indeed has a greater phenotypic value, we developed an estimator of the ratio between known and unknown effects on the phenotype. We evaluated prediction accuracy using human data from tens of thousands of individuals from either the same family or the same population, as well as data from different species. We found that, in many cases, even when only a small fraction of the loci affecting a phenotype is known, the individual with the greater phenotypic value can be identified with over 90% accuracy. Our approach also circumvents some of the limitations in transferring genetic association results across populations. Overall, we introduce an approach that enables accurate predictions of key information on phenotypes - the direction of phenotypic difference - and suggest that more phenotypic information can be extracted from genomic data than previously appreciated.
从基因组数据预测表型是遗传学的一个关键目标,但对于大多数复杂表型而言,预测因基因型到表型的映射不完整而受到阻碍。在此,我们描述了一种比定量预测更可行的方法,该方法旨在定性地预测表型差异。尽管基因型到表型的映射不完整,但我们表明确定两个个体中哪一个具有更大的表型值相对容易。这个问题在许多情形中都很关键,例如,比较个体之间的疾病风险、作物品系的产量,或已灭绝与现存物种的解剖结构。为了评估预测准确性,即预测表型较大的个体确实具有更大表型值的概率,我们开发了一种估计已知和未知表型效应之间比率的方法。我们使用来自同一家庭或同一群体的数万人的人类数据以及来自不同物种的数据来评估预测准确性。我们发现,在许多情况下,即使仅知道影响表型的一小部分基因座,也能够以超过90%的准确率识别出具有更大表型值的个体。我们的方法还规避了跨群体转移遗传关联结果时的一些局限性。总体而言,我们引入了一种能够准确预测表型关键信息——表型差异方向——的方法,并表明可以从基因组数据中提取比之前认识到的更多的表型信息。