Chen Denghui, Chitre Apurva S, Nguyen Khai-Minh H, Cohen Katarina, Peng Beverly, Ziegler Kendra S, Okamoto Faith, Lin Bonnie, Johnson Benjamin B, Sanches Thiago M, Cheng Riyan, Polesskaya Oksana, Palmer Abraham A
Bioinformatics and System Biology Program, University of California San Diego, 9500 Gilman Dr, La Jolla, CA 92093.
Department of Psychiatry, University of California San Diego, 9500 Gilman Dr, La Jolla, CA 92093.
bioRxiv. 2024 Jul 18:2024.07.17.603984. doi: 10.1101/2024.07.17.603984.
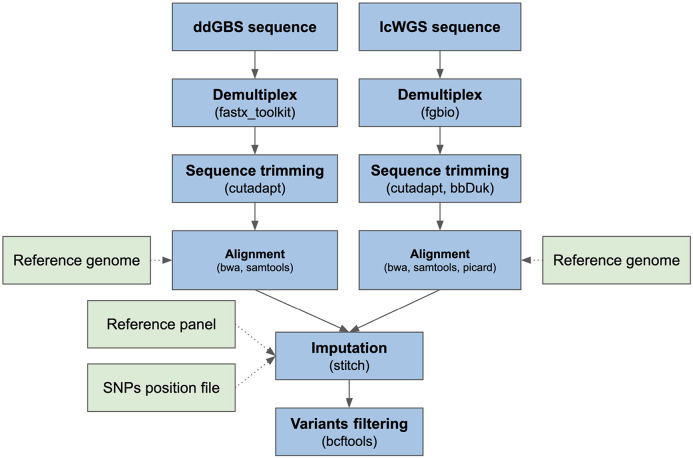
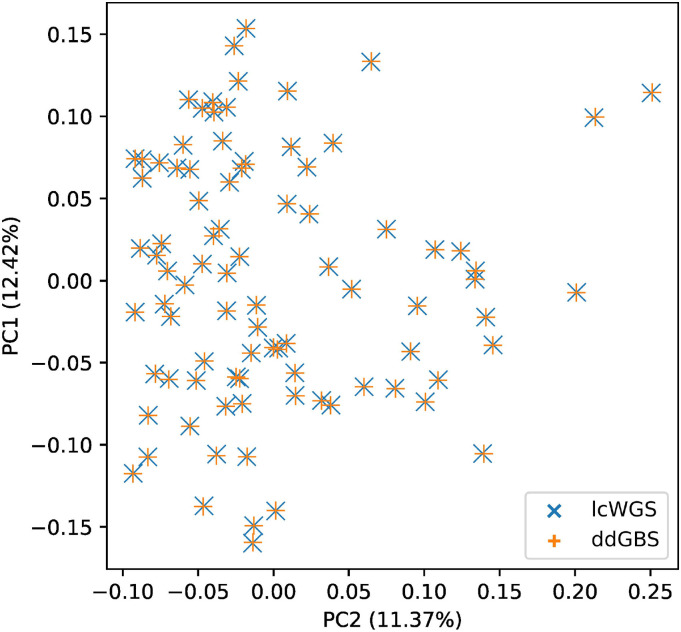
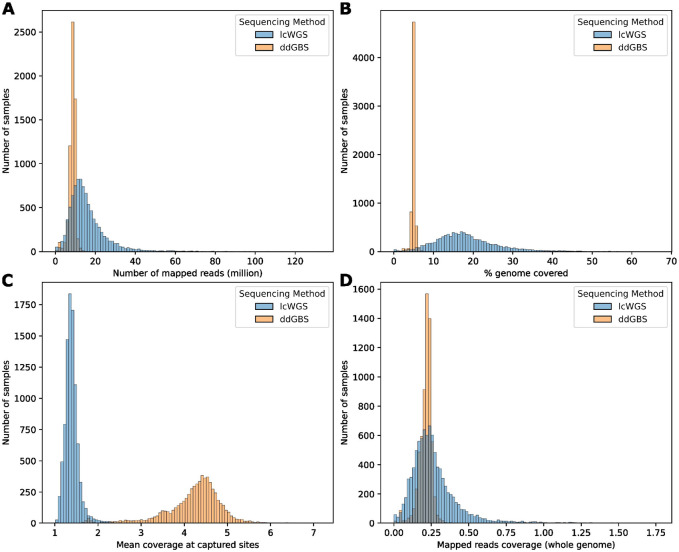
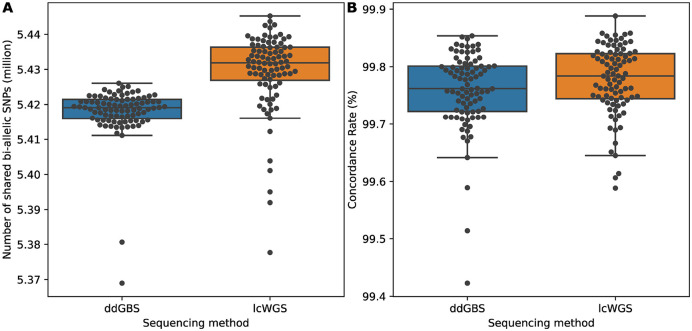
Affordable sequencing and genotyping methods are essential for large scale genome-wide association studies. While genotyping microarrays and reference panels for imputation are available for human subjects, non-human model systems often lack such options. Our lab previously demonstrated an efficient and cost-effective method to genotype heterogeneous stock rats using double-digest genotyping-by-sequencing. However, low-coverage whole-genome sequencing offers an alternative method that has several advantages. Here, we describe a cost-effective, high-throughput, high-accuracy genotyping method for N/NIH heterogeneous stock rats that can use a combination of sequencing data previously generated by double-digest genotyping-by-sequencing and more recently generated by low-coverage whole-genome-sequencing data. Using double-digest genotyping-by-sequencing data from 5,745 heterogeneous stock rats (mean 0.21x coverage) and low-coverage whole-genome-sequencing data from 8,760 heterogeneous stock rats (mean 0.27x coverage), we can impute 7.32 million bi-allelic single-nucleotide polymorphisms with a concordance rate >99.76% compared to high-coverage (mean 33.26x coverage) whole-genome sequencing data for a subset of the same individuals. Our results demonstrate the feasibility of using sequencing data from double-digest genotyping-by-sequencing or low-coverage whole-genome-sequencing for accurate genotyping, and demonstrate techniques that may also be useful for other genetic studies in non-human subjects.
经济实惠的测序和基因分型方法对于大规模全基因组关联研究至关重要。虽然人类受试者有基因分型微阵列和用于填充的参考面板,但非人类模型系统往往缺乏这些选择。我们实验室之前展示了一种使用双酶切测序基因分型对异质品系大鼠进行基因分型的高效且经济有效的方法。然而,低覆盖度全基因组测序提供了一种具有多种优势的替代方法。在此,我们描述了一种针对N/NIH异质品系大鼠的经济高效、高通量、高精度的基因分型方法,该方法可以结合先前通过双酶切测序基因分型生成的测序数据以及最近通过低覆盖度全基因组测序数据生成的数据。利用来自5745只异质品系大鼠的双酶切测序基因分型数据(平均覆盖度0.21x)和来自8760只异质品系大鼠的低覆盖度全基因组测序数据(平均覆盖度0.27x),与同一部分个体的高覆盖度(平均覆盖度33.26x)全基因组测序数据相比,我们可以填充732万个双等位基因单核苷酸多态性,一致性率>99.76%。我们的结果证明了使用双酶切测序基因分型或低覆盖度全基因组测序的测序数据进行准确基因分型的可行性,并展示了可能对非人类受试者的其他遗传研究也有用的技术。