Department of Biomedical Informatics, School of Medicine, University of Colorado - Anschutz Medical Campus, Aurora, CO, USA.
Division of Environmental and Occupational Health Sciences, Department of Medicine, National Jewish Health, Denver, CO, USA.
Respir Res. 2024 Jul 30;25(1):289. doi: 10.1186/s12931-024-02919-7.
Sarcoidosis is a heterogeneous granulomatous disease with no accurate biomarkers of disease progression. Therefore, we profiled and integrated the DNA methylome, mRNAs, and microRNAs to identify molecular changes associated with sarcoidosis and disease progression that might illuminate underlying mechanisms of disease and potential biomarkers.
Bronchoalveolar lavage cells from 64 sarcoidosis subjects and 16 healthy controls were used. DNA methylation was profiled on Illumina HumanMethylationEPIC arrays, mRNA by RNA-sequencing, and miRNAs by small RNA-sequencing. Linear models were fit to test for effect of sarcoidosis diagnosis and progression phenotype, adjusting for age, sex, smoking, and principal components of the data. We built a supervised multi-omics model using a subset of features from each dataset.
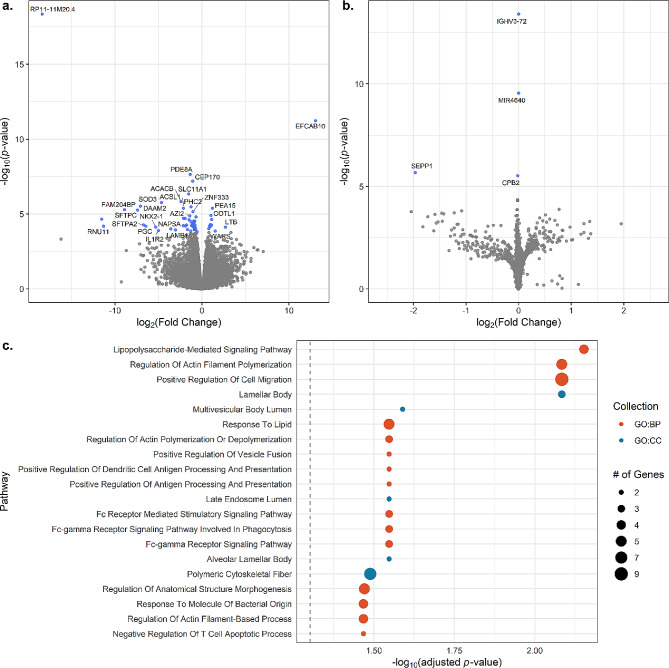
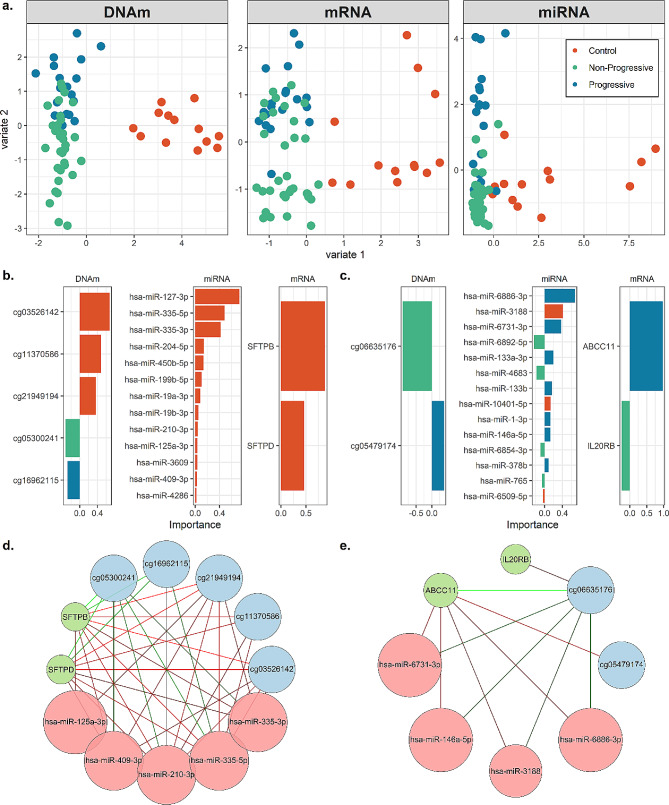
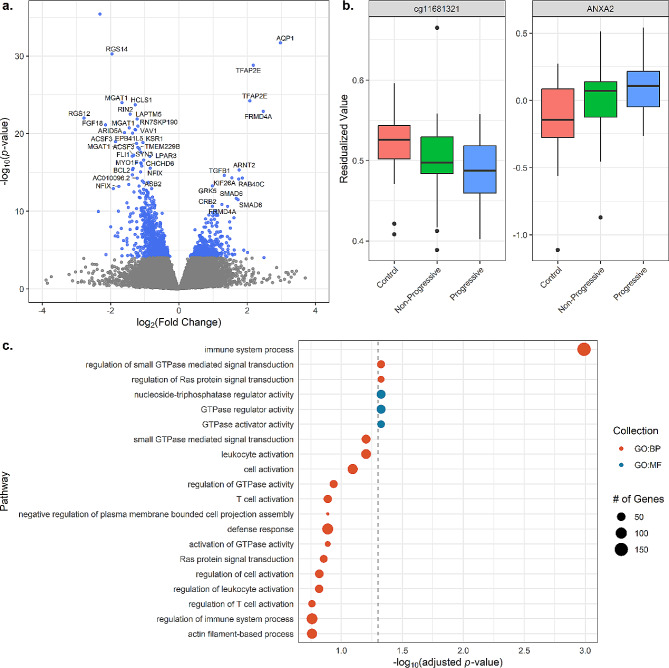
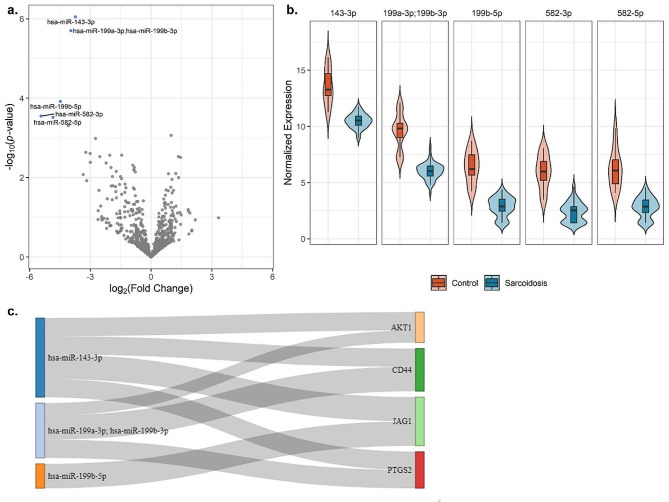
We identified 1,459 CpGs, 64 mRNAs, and five miRNAs associated with sarcoidosis versus controls and four mRNAs associated with disease progression. Our integrated model emphasized the prominence of the PI3K/AKT1 pathway, which is important in T cell and mTOR function. Novel immune related genes and miRNAs including LYST, RGS14, SLFN12L, and hsa-miR-199b-5p, distinguished sarcoidosis from controls. Our integrated model also demonstrated differential expression/methylation of IL20RB, ABCC11, SFSWAP, AGBL4, miR-146a-3p, and miR-378b between non-progressive and progressive sarcoidosis.
Leveraging the DNA methylome, transcriptome, and miRNA-sequencing in sarcoidosis BAL cells, we detected widespread molecular changes associated with disease, many which are involved in immune response. These molecules may serve as diagnostic/prognostic biomarkers and/or drug targets, although future testing is required for confirmation.
结节病是一种异质性肉芽肿性疾病,目前尚无疾病进展的准确生物标志物。因此,我们对 DNA 甲基化组、mRNA 和 microRNA 进行了分析和整合,以鉴定与结节病和疾病进展相关的分子变化,这些变化可能阐明疾病的潜在机制和潜在的生物标志物。
使用来自 64 例结节病患者和 16 例健康对照者的支气管肺泡灌洗液细胞。采用 Illumina HumanMethylationEPIC 芯片进行 DNA 甲基化分析,采用 RNA-seq 进行 mRNA 分析,采用 small RNA-seq 进行 miRNA 分析。通过线性模型检验结节病诊断和疾病进展表型的影响,调整年龄、性别、吸烟和数据的主成分。我们使用每个数据集的一部分特征构建了一个有监督的多组学模型。
我们鉴定出 1459 个 CpG、64 个 mRNA 和 5 个与对照组相比与结节病相关的 miRNA,以及 4 个与疾病进展相关的 mRNA。我们的整合模型强调了 PI3K/AKT1 途径的重要性,该途径在 T 细胞和 mTOR 功能中很重要。包括 LYST、RGS14、SLFN12L 和 hsa-miR-199b-5p 在内的新型免疫相关基因和 miRNA 可以区分结节病和对照组。我们的整合模型还显示,在非进展性和进展性结节病之间,IL20RB、ABCC11、SFSWAP、AGBL4、miR-146a-3p 和 miR-378b 的表达/甲基化存在差异。
在结节病 BAL 细胞中利用 DNA 甲基化组、转录组和 miRNA-seq,我们检测到与疾病相关的广泛分子变化,其中许多涉及免疫反应。这些分子可以作为诊断/预后生物标志物和/或药物靶点,但需要进一步的验证。